Manuel Perucho earned his PhD in biological sciences at the University of Madrid, Spain in 1976. He did postdoctoral work at the Max-Planck-Institut für Molekulare Genetik, Berlin and at Cold Spring Harbor Laboratory, where he was subsequently appointed to staff in 1981. Following appointments at SUNY Stony Brook as Assistant and Associate Professor in 1982 and 1987, respectively, Dr. Perucho joined the California Institute for Biological Research in La Jolla, serving as Research Program Director from 1993 to 1995. Dr. Perucho was recruited to Sanford Burnham Prebys in 1995.
Other Appointments
Adjunct Professor, Pathology Department, University of California, San Diego
Related Disease
Colorectal Cancer, Endometrial Cancer, Gastric Cancer, Ovarian Cancer
Dr. Perucho studies tumors from the intestinal tract that sometimes develop when the cellular machinery preserving the integrity of the genome – like computer spell-check programs that detect errors and correct them – is not working properly. When these corrector genes (mutators) are inactivated, the mutations that occur in all normal cells accumulate in large numbers because they are not repaired. This sparks genomic instability and cancer eventually develops when mutations occur in some cancer genes, such as oncogenes and tumor suppressor genes. However, some mutator genes are not inactivated by mutations, but by epigenetic silencing. This results from the disintegration of the epigenetic code, an unexplored process that is strongly associated with aging. This is important because many hereditary colon tumors originate by mutations in mutator genes that are transmitted from generation to generation. Molecular diagnosis of the deficient mutator genes determines which members of these families will be affected in the future. Identification of tumors with this kind of genomic instability is also useful for detecting familial cancer patients and predicting survival.
Manuel Perucho’s Research Report
Genomic Instability in Cancer Pathways
Our research efforts focus on the analysis of the genomic instability underlying two alternative pathways for oncogenesis (see figure below). Most neoplasms lose the chromosomal balance of the diploid normal cell following a pathway for cancer that involves the mutational inactivation of critical tumor suppressor genes. A minority of cancers manifest another type of genomic instability – the accumulation of hundreds of thousands of mutations, including insertions and deletions of a few base pairs in simple repeated sequences or microsatellites.
We study the aneuploidy of the tumor cell of the suppressor pathway for cancer by unbiased Arbitrarily Primed PCR DNA fingerprinting. Gains and losses of sequences from defined chromosomal regions can be simultaneously identified in multiple tumors generating a molecular karyotype or “amplotype.” Amplotyping offers useful applications for cancer diagnosis and prognosis and maps chromosomal regions harboring cancer genes with positive and negative roles in cell growth or survival. These cancer genes are under positive and negative selection pressure during tumorigenesis and are detected by the frequent gains and losses of specific chromosomal regions, respectively.
Understanding Colon Cancer
In colon cancer, the initial event in this carcinogenic pathway is the inactivation of the APC tumor suppressor. In hereditary cases of the familial polyposis (FAP) cancer syndrome, one mutated allele is transmitted in the germline, while in sporadic cases both alleles are inactivated by somatic mutations. Usually one allele is inactivated by a nonsense or frameshift mutation and the other allele is inactivated by the deletion of the chromosomal region (loss of heterozygosity), typical of the aneuploid cancer cell.
The first event in the Microsatellite Mutator Phenotype (MMP) pathway for colon cancer is the inactivation of a gene involved in genome stability, such as the hMLH1 DNA mismatch repair gene. In sporadic cases, the inactivation of the mutator gene usually occurs by somatic mutations or by epigenetic silencing. In many familial cases, including a majority of the Hereditary Non-Polyposis Colorectal Cancer (HNPCC) syndrome, one allele is inactivated by a germline mutation and the other by any of the other mechanisms (mutation, LOH, epigenetic silencing, etc.).
The MMP pathway for gastrointestinal cancer presents two distinctive features that seem paradoxical at first sight. First, despite accumulating hundreds of thousands of clonal somatic mutations in simple repeated sequences, these tumors exhibit a low mutation incidence in APC, K-ras and p53, prototypical cancer genes in colorectal carcinogenesis. Second, these tumors harbor ubiquitous biallelic mutations in non-functional poly (A)n sequences, such as the poly A tails of the Alu repeats. However, they also accumulate many monoallelic (i.e., heterozygous) mutations in functional sequences, such as the coding regions of mutator (hMSH3, hMSH6), suppressor (TGFbRII, p53) and apoptotic (Bax) genes.
The first paradox may be explained by the existence within some genes of simple repeats that are preferred targets for the MMP. Thus, in the presence of the mutator phenotype, mutations in these genes (i.e., Bax) occur sooner than in other genes of the same oncogenic signaling pathways that do not have these repeats (i.e., p53). The second paradox can also be explained by another peculiar feature of these MMP tumors. Due to their exacerbated mutator phenotype, the disruption of the homeostatic controls for cell growth and survival may also occur by the accumulation of heterozygous mutations in multiple genes whose products play redundant but synergistic roles at different points of the cell proliferation and apoptotic networks. The occurrence of multiple heterozygous mutations presumably reduces the threshold amounts of the corresponding gene products. This accumulative haploinsufficiency model is not restricted to cell proliferation and apoptotic pathways, but also applies to other networks involved in the control of genome integrity.
Minoru Fukuda earned his PhD in biochemistry from the University of Tokyo in 1973 and did his postdoctoral training at the Yale University School of Medicine. Following a period with joint appointments at University of Washington and Fred Hutchinson Cancer Research Center in Seattle, he was recruited to Sanford-Burnham Medical Research Institute in 1982 as Director of the Glycobiology Program. Dr. Fukuda directs the program project grant, which consolidates the research efforts of the members of the Glycobiology Program.
Dr. Fukuda is a recipient of a Merit Award from the National Cancer Institute and the 1997 recipient of the Karl Meyer Award from the Society of Glycobiology. He served as an Executive Editor for Biochimica et Biophysica Acta, as an Associate Editor for Cancer Research and Editorial Member for Journal of Biological Chemistry. He also has edited 11 books including three books from Oxford University Press and three volumes of Methods in Enzymology and holds an Adjunct Professor appointment at the University of California, San Diego.
Education
1973: PhD, University of Tokyo, Biochemistry 1970: MS, University of Tokyo, Biochemistry 1968: BS, University of Tokyo, Biochemistry
Related Disease
Brain Cancer, Colorectal Cancer, Gastric Cancer, Helicobacter pylori, Prostate Cancer
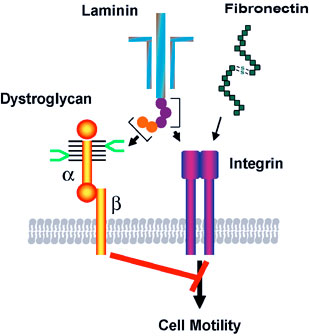
The cell surface is heavily coated with carbohydrates. The structure of those cell surface carbohydrates displays a dramatic change during development, and mature cells express cell surface carbohydrates specific to different organs and tissues. Cell surface carbohydrates thus serve as a zip code for different organs and tissues. Sialyl Lewis X represents such an oligosaccharide. After discovery of sialyl Lewis X in neutrophils by Dr. Minoru Fukuda, his laboratory demonstrated that sulfated form of sialyl Lewis X is essential for lymphocyte homing and recruitment of natural killer cells in preventing tumor metastasis to the peripheral lymph node. With colleagues from Japan, Dr. Fukuda discovered that certain carbohydrates function as antibiotics against Helicobacter pylori infection, which is a leading cause for peptic ulcer and gastric carcinoma. Most recent studies in Dr. Fukuda’s laboratory revealed that decrease of the laminin-binding glycans on α-dystroglycan in carcinoma cells leads to tumor cell migration, invasion, and metastasis. The restoration of the unique glycans by the expression of distinct β3-N-acetylglucosaminyltransferase renders these cells act like normal cells. The results indicate that certain carbohydrates on normal cells and enzymes that synthesize those glycans, such as β3-N-acetylglucosaminyltransferase, function as tumor suppressors These findings will be useful in developing carbohydrate-based therapy for the treatment of inflammation and tumor metastasis.
Minoru Fukuda’s Research Report
Cell Surface Carbohydrates as Tumor Suppressor
Many studies have focused on carbohydrates that increase in cancer cells, but only a few have looked at carbohydrates that appear in normal cells but decrease or disappear in cancer cells. A specific mucin-type O-glycans (core 3 O-glycans) is one of such glycans, and we found core 3 O-glycans suppress tumor formation and metastasis. When core 3 O-glycans were forced to express on human prostate cancer cell lines, those prostate cancer cells produced much smaller tumors and almost no metastasis. By contrast, the parent cancer cells, which did not express core 3 O-glycans, produced robust primary and metastatic tumors. We showed that the expression of core 3 O-glycans decreases a formation of α2β1-integrin complex, receptors that mediate cell adhesion, diminishing cancer cell migration.
We also revealed tumor suppressor function in the unique laminin-binding glycans on dystrophin complex, — carbohydrates located on α-dystroglycan, which is also associated with cell adhesion. We discovered that the unique glycans play a critical role in epithelial-basement membrane interaction in normal cells, and the decrease or loss of the glycans, due to downregulation of β3-N-acetylglucosaminyltransferase, leads to increased malignancy by invasive carcinoma cells. Restoration of the laminin-binding glycans by forced expression of β3-N-acetylglucosaminyltransferase, on the other hand, results in reduced cell migration, thus dramatic decrease in tumor formation and metastasis. We demonstrated that interaction of laminin with the unique glycans on α-dystroglycan counteracts the cell migration signals that are mediated by integrin binding to its ligands, thereby decreasing tumor formation and metastasis. These findings also suggest that the laminin-binding glycans can be an excellent marker for epithelial-mesenchymal transitions.
These results indicate that certain carbohydrates on normal cells and enzymes that synthesize those glycans, such as β3-N-acetylglucosaminyltransferase, function as tumor suppressors. Upregulation of those key enzymes may become a novel way to treat cancer.
Neural Cell-specific Glycans in Development and Cancer
Polysialic acid and HNK-1 glycan represent carbohydrates enriched in neural cells. Polysialic acid is mainly attached to NCAM in embryos, while the majority of NCAM in adults lack this carbohydrate. To understand the roles of these glycans in neural development, we have cloned cDNAs encoding human polysialyltransferase, PST, and HNK-1 sulfotransferase, HNK-1ST, that are responsible for the synthesis of polysialic acid and HNK-1 glycan, respectively. By using these cloned cDNAs, we demonstrated that polysialic acid facilitates the invasion of glioma, the most common form of adult brain tumor. Our studies also showed that mutant mice with deficient STX, another polysialyltransferase, exhibit reduced behavioral response to fear conditioning, apparently due to anomalies in mossy fibers of the hippocampus. Our studies also demonstrated that neural development is significantly impaired in mutant mice that entirely lack polysialic acid due to inactivation of two polysialyltransferases. We found that this defect is caused by impairment of neural cell migration.
Mucin-type O-glycans in Immune Cell Interactions
Previously, we found that an increase of core 2-branched oligosaccharides is associated with leukemia and immunodeficiency, such as in Wiskott-Aldrich syndrome and AIDS. To determine the roles of core 2-branched O-glycans in immune cell interactions, the enzyme (C2GnT-1) responsible for the core 2-branched oligosaccharide was knocked-out by gene targeting. Compared to wild-type mice, leukocytes from the gene knockout mice exhibited a reduced binding to L-, E- and P-selectin in this order. In contrast, homing of lymphocytes was moderately reduced. Lymphocyte homing is mediated by binding of L-selectin on lymphocytes to sulfated L-selectin oligosaccharide ligands, 6-sulfo sialyl Lewis X in high endothelial venules (HEV) of secondary lymphoid organs. By analyzing remaining L-selectin ligands in C2GnT-1 knockout mice, we discovered novel L-selectin ligands that are based on extended core 1 oligosaccharides. The core portion of this novel L-selectin ligand is also an epitope for MECA-79 antibody that inhibits lymphocyte homing in vivo. Moreover, crossbreeding between mutant mice with deficient L-selectin ligand sulfotransferase and another sulfotransferase led to our findings that these two enzymes in cooperation synthesize L-selectin ligands. These mutant mice lack 6-sulfate group in L-selectin ligands that results in impaired inflammatory response. More recently, mice deficient in L-selectin ligands on mucin-type O-glycans were generated. The studies on the mutant mice revealed novel functions of N-glycan-based L-selectin ligand, which supports both lymphocyte homing and inflammatory response. This finding brought a new paradigm in selectin-carbohydrate interaction.
Carbohydrate-dependent Adhesion in Tumor Metastasis
Previously we found that the amount of core 2 O-glycans is significantly increased in colon and lung carcinomas and the increase of core 2 O-glycans is highly correlated to vessel invasion and lymph node metastasis. More recently, we discovered that forced expression of core 2 O-glycans by transfecting C2GnT-1 in prostate cancer cell lines resulted in increased tumor formation.
In parallel, we discovered that forced expression of selectin ligands, sialyl Lewis X on B16 melanoma cells leads to increased lung tumor formation. We also showed that tumor formation in lymph node is suppressed by natural killer (NK) cells, which are recruited by L-selectin mediated homing of NK cells to lymph nodes.
Roles of Carbohydrates in Helicobacter pylori-mediated Inflammation and Cancer
Helicobacter pylori is a leading cause of peptic ulcer and gastric cancer. Previously it was shown by others that H. pylori adhere to gastric mucosa in a carbohydrate-dependent manner. The infection of H. pylori leads to chronic inflammation, which apparently leads to peptic ulcer and gastric cancer.
Our recent studies showed that H. pylori-induced inflammation is associated with the formation of peripheral lymph node addressin (PNAd) characterized by binding to MECA-79 antibody and L-selectin. The number of HEV-like vessels expressing PNAd increases as H. pylori-induced inflammation progresses. Moreover, PNAd disappears once H. pyloriis eradicated by antibiotic treatment. These findings indicate that H. pylori-induced inflammation is facilitated by de novo formation of PNAd thereby recruiting lymphocytes. It may be possible to attenuate or prevent the formation of peptic ulcers or gastric cancer by inhibiting L-selectin ligand synthesis, for example by inhibiting the sulfotransferases.
Binding of laminin to the specific carbohydrate (shown in bright green) on α-dystroglycan counteracts the migration signals initiated by integrin binding to extracellular matrix proteins such as laminin and fibronectin. The synthesis of this specific carbohydrate requires a unique β3-N-cetylglucosaminyltransferase, and the downregulation of the glycosyltransferase in carcinoma cells leads to increased cell migration, thereby increased tumor formation and metastasis. Thus the specific carbohydrate structure at cell surface functions as a tumor suppressor, which is controlled by the unique β3-N-acetylglucosaminyltransferase.
While over half of the world’s population is infected with H. pylori, only a fraction of those individuals progress to peptic ulcer and gastric cancer. In relation to these observations, α1,4-N-acteylglucosaminyl capping structure (α4GlcNAc) is present in deeper portions of the gastric mucosa, where H. pylori rarely colonizes. We discovered that α4GlcNAc capping structure functions as an antibiotic against H. pylori infection by inhibition of the synthesis of α-glucosyl cholesterol, a major component of the H. pyloricell wall. This unprecedented discovery should be useful in developing drugs to inhibit H. pylori colonization, through inhibition of cholesterol α-glucosyltransferase. Such drugs lead to a novel treatment for prevention and potential treatment of peptic ulcer and gastric carcinoma.
Pamela Itkin-Ansari earned her PhD in Biomedical Sciences from the University of California, San Diego, in 1999. She received postdoctoral training focused on diabetes at that same organization. In 2003, Dr. Itkin-Ansari was appointed Assistant Professor in the Department of Pediatrics, UC San Diego. She moved her laboratory to Sanford Burnham Prebys in 2005.
2013-2014: Outstanding Faculty Mentor Award, UCSD 2011-2014: Hartwell Foundation Biomedical Research Award 2011: Invited Speaker, TEDx 2008-2013: Board of Directors, JDRF San Diego 2008: Health Hero Leadership Award, Combined Health Agencies of San Diego
Other Affiliations
2012-current: Islet Society 2010-current: ASGCT 2008-current: Board of Directors, JDRF San Diego chapter 2008-2013: JDRF board of directors, San Diego 2007-current: American Association for Cancer Research 2007-current: American Diabetes Association 2007-current: American Pediatric Society/Society for Pediatric Research 2006-current: AAAS
Related Disease
Cancer, Diabetes – General, Gastric Cancer, Monogenic Diabetes, Pancreatic Cancer, Type 1 Diabetes, Type 2 Diabetes
Dr. Itkin-Ansari’s research is directed toward understanding diseases of the human pancreas.
Pamela Itkin-Ansari’s Research Report
Diabetes
Areas of focus are: 1) developing a cell-based therapy for diabetes that does not require immunosuppression, and 2) identifying proteins required for proper insulin production and processing.
Islet clusters in the developing pancreas
Pancreatic Cancer
The lab determined how dysregulation of specific transcription factors triggers pathogenic cell cycle entry in normal pancreatic cells. This master signaling pathway controlling pancreatic cancer cell growth is yielding potential targets for drug discovery.