Dr. Piña-Crespo earned a PhD in Pharmacology from University College London (UCL), England under the supervision of Profs. Alasdair Gibb & David Colquhoun FRS. He completed postdoctoral training as a Pew Fellow/Research Associate with Prof. Steve Heinemann in the Molecular Neurobiology Laboratory at The Salk Institute, La Jolla, California. Dr. Piña-Crespo has held faculty positions as Instructor and Assistant Professor at Universidad Centroccidental, Venezuela and as Lecturer in the Biology Department at the University of San Diego, California.
Education and Training
Postdoctoral training (Pew Fellow/Research Associate) The Salk Institute, California PhD in Pharmacology University College London (University of London), England Veterinarian (DVM) Universidad Centroccidental Lisandro Alvarado, Venezuela
Honors and Recognition
Pew Fellow in the Biomedical Sciences
Related Disease
Aging-Related Diseases, Alzheimer’s Disease, Brain Injury, Epilepsy, Molecular Biology, Nervous System Injury, Neurodegenerative and Neuromuscular Diseases, Neurological and Psychiatric Disorders, Parkinson’s Disease, Stroke, Traumatic Injury
Phenomena or Processes
Aging, Apoptosis and Cell Death, Calcium Signaling, Cell Biology, Cell Signaling, Cell Surface Receptors, Development of Neuronal Circuits, Disease Therapies, Neurobiology, Neurogenesis, Neuron-Glia Interactions in Myelin, Neurotransmitters, Synapse Function, Synaptic Transmission
Anatomical Systems and Sites
Brain, General Cell Biology, Nervous System
Research Models
Cultured Cell Lines, Human Cell Lines, Human Embryonic Stem Cells, Mouse, Mouse Cell Lines, Primary Cells, Primary Human Cells, Rat, Vertebrates, Xenopus
Techniques and Technologies
Biophysics, Biophysiology, Calcium Imaging, Cellular and Molecular Imaging, Electrophysiology, Fluorescence Microscopy, Ion Channels, Live Cell Imaging, Mouse Behavioral Analysis, Pharmacology, Transplantation
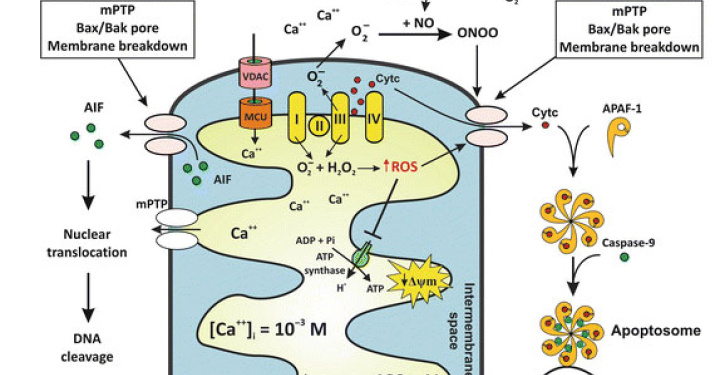
Working on basic neuroscience discovery research. I use cellular and animal models of neurodegeneration to identify basic disease-causing mechanisms and disease-relevant targets involved in abnormal neuron-glia signaling, synapse failure, neuronal network dysfunction and neuronal loss in age-related neurodegenerative diseases; including Alzheimer’s and Parkinson’s disease. Extensive hands-on experience working and managing projects that require a strong background in in-vitro, ex-vivo and in-vivo neuroscience, pharmacology and electrophysiology.
Anatomical Systems and Sites
Immune System and Inflammation
Research Models
Clinical and Transitional Research, Computational Modeling, Human, Human Cell Lines, Mouse, Mouse Cell Lines, Primary Cells, Primary Human Cells
Techniques and Technologies
Biochemistry, Bioinformatics, Cellular and Molecular Imaging, Drug Discovery, Drug Efficacy, Gene Expression, Gene Knockout (Complete and Conditional), Gene Silencing, High-Throughput/Robotic Screening, RNA Interference (RNAi), Systems Biology
Eric has a broad background in chemical biology, with specific training and expertise in kinase inhibitors and targeted protein degradation, an emerging modality in which small molecules recruit E3 ligase complexes to target proteins to induce their ubiquitination and subsequent proteasomal degradation. He also has experience in pharmacological modulation of immune cells to improve anti-tumor immunity.
He received his PhD from the University of California San Francisco and postdoctoral training at the Dana-Farber Cancer Institute.
Education and Training
2021: Postdoctoral Fellow, Dana-Farber Cancer Institute / Harvard Medical School 2015: PhD, University of California San Francisco 2009: BS, Duke University
Fellowship
Damon Runyon Cancer Research Foundation Fellowship
Related Disease
Cancer, Immune Disorders, Molecular Biology
Phenomena or Processes
Adaptive Immunity, Cancer Biology, Cell Biology, Disease Therapies, Tumor Microenvironment
Techniques and Technologies
Chemical Biology, Drug Discovery
Dysregulation of transcriptional circuits is a common hallmark of disease, and in particular is found in both tumor and host immune cells in cancer. Eric’s lab is focusing on developing and using chemical tools to modulate the activity of key transcriptional regulators of both tumor cells and host immune cells, with a long-term goal of identifying new therapeutic approaches.
Ten scientists at Sanford Burnham Prebys Medical Discovery Institute were awarded eight grants yesterday from Curebound, a San Diego-based philanthropic organization
Charles Spruck earned his BS in Biology at UCLA and PhD in Molecular Biology at the University of Southern California. He worked as a postdoctoral fellow at The Scripps Research Institute in La Jolla and was recruited to the Sidney Kimmel Cancer Center in San Diego as an Assistant Professor in 2003. He joined Sanford Burnham Prebys in 2010.
Education and Training
2003: Post-doc, The Scripps Research Institute 1986: PhD, University of Southern California 1995; BS, University of California at Los Angeles
Prestigious Funding Awards / Major Collaborative Grants
NIH/NCI DoD BCRP CBCRP TRDRP
Honors and Recognition
ACS Scholar
Related Disease
Breast Cancer, Cancer, Lung Cancer, Molecular Biology
Phenomena or Processes
Cancer Biology, Cancer Epigenetics, Cell Biology, Cell Cycle Progression, Cell Signaling, Genomic Instability, Innate Immunity, Metastasis, Posttranslational Modification, Proteolytic Pathways
Research Models
Cultured Cell Lines, Human Cell Lines, Mouse, Mouse Cell Lines
Techniques and Technologies
Cell Biology, Drug Discovery, Gene Knockout (Complete and Conditional), In vivo Modeling
“Despite recent advances in treatment, patients with advanced metastatic cancers have few treatment options. Our lab is focused on developing new effective and non-toxic treatments for these patients.”
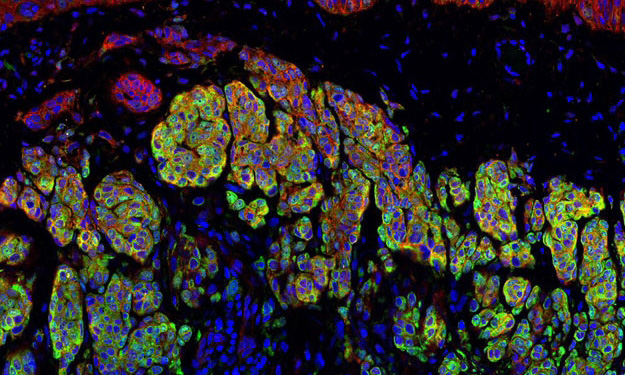
Dr. Spruck’s laboratory is focused on developing new, effective, and non-toxic treatments for patients with advanced cancers. The lab focuses on defining the molecular networks that regulate cancer cell division and drive metastasis progression. Recent studies have focused on viral mimicry as a therapeutic approach in cancer, which involves the activation of dormant endogenous retroviruses and retrotransposons in cancer cells to enhance immunogenicity and the effectiveness of immune checkpoint blockade immunotherapy and DNA damaging therapies. The laboratory utilizes various biochemical and molecular approaches, CRISPR gene editing, and animal models of cancer. An emphasis is on studies of breast, lung, prostate, and brain tumors.
Charles Spruck’s Research Report
Developing viral mimicry therapeutic approaches for cancer: Approximately 45% of the human genome is composed of repetitive elements (REs), including endogenous retroviruses and retrotransposons, that are normally transcriptionally silenced in somatic cells. Recent studies suggest that the transcriptional awakening of ERVs/retrotransposons beyond a threshold level of tolerance in cancer cells induces antiviral responses that can enhance the efficacy of certain therapies, including immunotherapy. We recently discovered a novel epigenetic regulatory pathway, FBXO44/SUV39H1, that is essential for ERV/retrotransposon silencing in cancer cells. Preclinical studies showed that FBXO44/SUV39H1 inactivation induces viral mimicry in cancer cells, leading to increased immunogenicity, decreased tumorigenicity, and enhanced the efficacy of immune checkpoint blockade therapy. We are currently exploring therapeutic approaches to target this pathway, and others like it, to prevent tumor growth and enhance immunotherapy response. We are also exploring the role of reactivated REs in human diseases.
Targeting metastatic tumors: Metastasis is a major cause of mortality in cancer. Through genomic screens and biochemical studies, we are identifying novel molecular pathways that drive cancer cell motility, invasion, and metastasis. Recently, we identified a novel molecular axis, FBXO7/EYA2-SCF(FBXW7), that promotes cancer cell motility and cancer stem cell self-renewal and suppresses cancer cell immunogenicity. Targeting this axis prevented metastasis progression, reduced the cancer stem cell population, and stimulated anti-tumor immune responses in preclinical mouse breast cancer models.
Ten scientists at Sanford Burnham Prebys Medical Discovery Institute were awarded eight grants yesterday from Curebound, a San Diego-based philanthropic organization
Pier Lorenzo Puri earned his MD at the University of Rome “la Sapienza” in 1991. Dr. Puri completed his internship in Internal Medicine at the hospital “Policlinico Umberto I” (Rome) from 1992 to 1997, and defended an experimental thesis on the vascular effects of angiotensin II to graduate as Specialist in Internal medicine at the University of Rome “la Sapienza” in 1997. During this time he was frequently working at the Freien University of Berlin, as visiting scientist at the Deprtment of Biochemistry and Molecular Biology, to perform experiments of protein and DNA microinjection in cultured cells. Dr. Puri trained as a post-doctoral fellow at the University of California San Diego (UCSD), in the department of Cell Biology, under the supervision of Dr. Wang, from 1997 to 2001. He was appointed as Staff Scientist at the Salk Institute (La Jolla) in 2001, and became an Assistant Telethon Scientist at the Dulbecco Telethon Institute in Rome in 2002. He was upgraded to Associate Telethon Scientist at the Dulbecco Telethon Institute in Rome since 2007 and became Senior Telethon Scientist, Dulbecco Telethon Institute, in 2012, but declined this position. Dr. Puri joined Sanford Burnham Prebys as an Assistant Professor in 2004. He has been promoted to Associate Professor in 2010 and full Professor in 2015. From 2008 to 2016 Dr. Puri served as Adjunct Professor of Pediatrics at the University of California, San Diego. From 2008 to 2013 Dr Puri was an Associate Member of Sanford Children’s Health Research Center. Dr Puri has been Director of the laboratory of Epigenetics and Regeneration at Fondazione S. Lucia, Roma, Italy, but stepped down this position since 2019.
Education
University of California San Diego, Postdoctoral, Department of Biology University of Rome La Sapienza, PhD, Internal Medicine University of Rome La Sapienza, MD, Internal Medicine University of Rome La Sapienza, Undergraduate, Internal Medicine
Other Appointments
2020-2024: Member of the Science Advisory Board (SAB) European Commission-funded Consortium BIND (Brain Involvement In Dystrophinopathies) 2015-2019: Standing Member, NIH Study Section (SMEP) 2010-present: Member of Editorial Board of Skeletal Muscle
Phenomena or Processes
Adult/Multipotent Stem Cells, Aging, Cell Biology, Cell Cycle Progression, Cell Differentiation, Cell Signaling, Cellular Senescence, Development and Differentiation, Disease Therapies, DNA Damage Checkpoint Function, Epigenetics, Gene Regulation, Phosphorylation, Regenerative Biology, Signal Transduction, Transcriptional Regulation
Anatomical Systems and Sites
General Cell Biology, Musculoskeletal System
Research Models
Clinical and Transitional Research, Cultured Cell Lines, Human Adult/Somatic Stem Cells, Mouse Embryonic Stem Cells, Mouse Somatic Stem Cells, Primary Human Cells
Techniques and Technologies
Bioinformatics, Cellular and Molecular Imaging, Gene Expression, Genomics
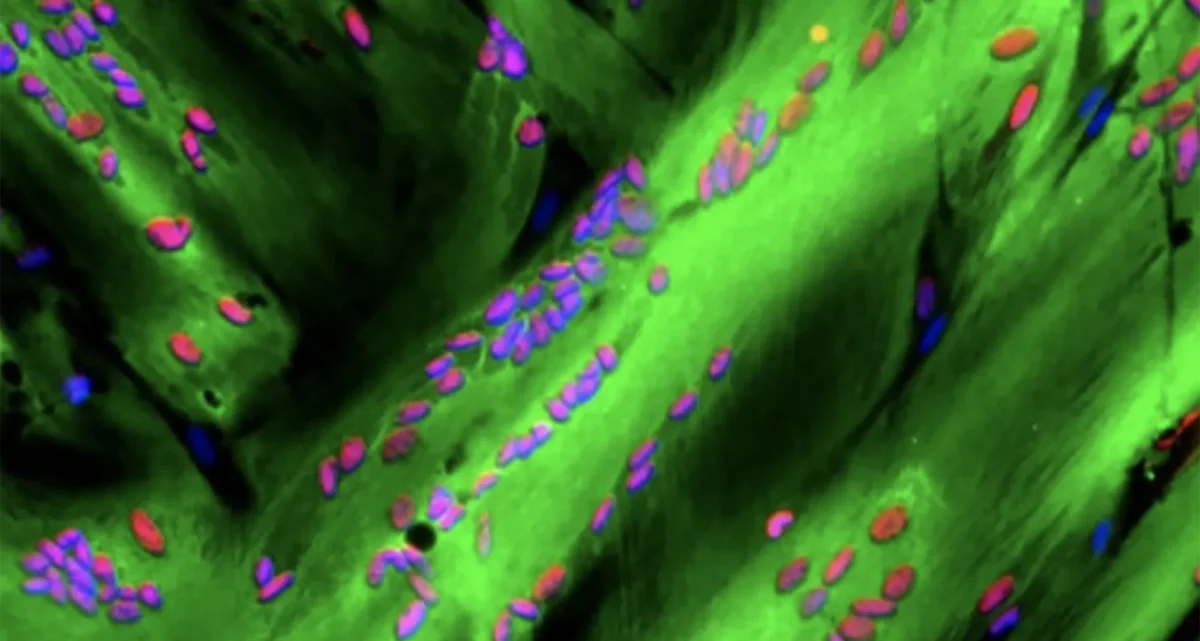
Puri’s lab group investigates the molecular and epigenetic regulation of gene expression in skeletal muscle progenitors and other muscle-resident cell types (including fibro-adipogenic progenitors, cells from the inflammatory infiltrate, cellular components of neuro-muscular junctions) during physiological and pathological perturbations of skeletal muscle homeostasis.
We use molecular, biochemical and epigenetic tools to understand structural and functional principles of the 3D genome organization that regulates gene expression during muscle regeneration and diseases.
A topic of particular interest is the analysis of chromatin interactions that define the 3D genome organization and the identification of structural and functional interactions that regulate cell type-specific patterns of gene expression in response to cues released within the skeletal muscle regenerative environment in health and disease conditions, such as muscular dystrophies and other neuromuscular diseases.
The knowledge derived from our studies is instrumental to elucidate the pathogenesis of muscular disorders and discover pharmacological interventions that promote muscle regeneration to repair diseased muscles.
Current translational focus is devoted to:
the study of the therapeutic potential of HDAC inhibitors for treatment of Duchenne Muscular Dystrophy (DMD)
the identification of genome variants associated to DMD patient-specific patterns of expression of disease-modifier genes that can account for individual trends of disease progression beyond the common genetic deficiency of dystrophin
the effect of dystrophin deficiency and restoration by gene therapy on 3D genome and transcriptional output of DMD myofibers; the therapeutic potential of extracellular vesicles released by fibro-adipogenic progenitors of DMD skeletal muscles exposed to HDACi.
Puri Lab
Pier Lorenzo Puri’s Research Report
Puri’s lab group investigates the molecular and epigenetic regulation of gene expression in skeletal muscle progenitors and other muscle-resident cell types (including fibro-adipogenic progenitors, cells from the inflammatory infiltrate, cellular components of neuro-muscular junctions) during physiological and pathological perturbations of skeletal muscle homeostasis.
We use molecular, biochemical and epigenetic tools to understand structural and functional principles of the 3D genome organization that regulates gene expression during muscle regeneration and diseases
A topic of particular interest is the analysis of chromatin interactions that define the 3D genome organization and the identification of structural and functional interactions that regulate cell type-specific patterns of gene expression in response to cues released within the skeletal muscle regenerative environment in health and disease conditions, such as muscular dystrophies and other neuromuscular diseases.
The knowledge derived from our studies is instrumental to elucidate the pathogenesis of muscular disorders and discover pharmacological interventions that promote muscle regeneration to repair diseased muscles
Current translational focus is devoted to:
the study of the therapeutic potential of HDAC inhibitors for treatment of Duchenne Muscular Dystrophy (DMD)
the identification of genome variants associated to DMD patient-specific patterns of expression of disease-modifier genes that can account for individual trends of disease progression beyond the common genetic deficiency of dystrophin
the effect of dystrophin deficiency and restoration by gene therapy on 3D genome and transcriptional output of DMD myofibers; the therapeutic potential of extracellular vesicles released by fibro-adipogenic progenitors of DMD skeletal muscles exposed to HDACi.
1. Epigenetic regulation of skeletal myogenesis by histone acetyltransferases and deacetylases
Our earlier identification and characterization of acetyltransferases p300/CBP and PCAF, as transcriptional co-activators, and the histone deacetylases HDACs, as transcriptional co-repressors, of the myogenic determination factor MyoD1-3, respectively, inspired the experimental rationale toward exploiting pharmacological inhibition of HDAC to promote skeletal myogenesis.
2. HDAC inhibitors as pharmacological intervention in DMD and other muscular dystrophies
Puri lab discovered that dystrophin-activated nNOS signalling controls HDAC2 activity, thereby revealing a previously unrecognized link between constitutive activation of HDAC2 and alteration of the epigenetic landscape of dystrophin-deficient muscles6,7. This discovery established the rationale for using HDAC inhibitors to counter the progression of Duchenne muscular dystrophy (DMD), by correcting aberrant HDAC activity in dystrophin-deficient muscles8-11.
3. Control of chromatin structure in muscle cells by regeneration-induced signaling pathways
Upon the discovery and characterization of intracellular signaling pathways (i.e. p38, ERK and AKT cascades) that regulate muscle gene expression in myoblasts, in earlier studies during Puri’s postdoctoral training, Puri lab has revealed the mechanism by which muscle environmental cues are converted into epigenetic changes that regulate gene expression in healthy and diseased muscles, via extracellular signal-activated kinase targeting of chromatin-modifying enzymes. These studies provided the first evidence that regeneration activated p38 and AKT signaling cooperatively direct assembly and activation of histone acetyltransferases and chromatin remodeling SWI/SNF complex at myogenic loci in muscle progenitors12,13,15. Moreover, we discovered that regeneration-activated p38 targets Polycomb Repressory Complex (PCR2) at Pax7 locus to promote formation of repressive chromatin during satellite cells a ctivation14.
4. Epigenetic basis for activation of the myogenic program in ESCs and other pluripotent cell types
Puri lab studied the epigenetic determinants of human embryonic stem cells (hESCs) and induced pluripotent stem cells (hiPSCs) commitment to skeletal myogenesis, by investigating the hESC resistance to direct conversion into skeletal muscle upon ectopic expression of MyoD, which can otherwise reprogram somatic cells into the skeletal muscle lineage. These studies showed that hESC and hiPSC resistance to myogenic conversion is caused by the lack of expression of one structural component of the SWI/SNF chromatin remodelling complex – BAF60C – which is specifically induced in embryoid bodies13. Based on these studies, we have recently established a protocol of hESC-derived 3D contractile myospheres that offers the unprecedented opportunity to dissect and analyze the epigenetic dynamics that underlie the formation of skeletal muscles and to identify changes in the epigenome induced by contractile activity in healthy vs dystrophin-deficient myofibers16,20. We have also determined the identity of the general transcription factors implicated in the activation of skeletal myogenesis17, and we have discovered that replicative senescence is associated with acquisition of resistance to MYOD-mediated activation of muscle gene expression, caused by the constitutive activation of DNA damage repair (DDR) response that impairs cell cycle progression and MYOD activity18. Finally, our recent work has elucidated the mechanism by which MYOD regulates high-order chromatin interactions to define the tri-dimensional (3D) nuclear architecture for the activation of skeletal myogenesis during human somatic cell reprogramming into skeletal muscles19.
5. Identification, functional, phenotypic and molecular characterization of muscle-interstitial cells – (the fibroadipogenic progenitors – FAPs) in healthy and diseased muscles.
Our work has elucidated the molecular determinants of the interplay between adult muscle stem cells and cellular components of their functional niche (i.e. FAPs), by identifying regulatory networks implicated in compensatory or pathogenic regeneration, and suggesting “disease stage-specific” responses to pharmacological treatment of neuromuscular disorders, such as DMD. Indeed, we have shown that HDACi promote compensatory regeneration and prevent fibro-adipogenic degeneration in mdx mice at early stages of diseases, by targeting a population of muscle interstitial cells – FAPs8 – and have identified a HDAC-regulated network that controls expression of myomiRs and alternative incorporation of BAF60 variants into SWI/SNF complexes to direct the pro-myogenic or fibro-adipogenic FAP activity21. Furthermore, we have recently identified specific subpopulations of FAPs (subFAPs) in physiological conditions and disease22 and we have discovered that specific subFAPs expand and adopt pathogenic phenotypes upon muscle denervation23 or in muscles of patients affected by type 2 diabetes24.
After a successful teaching career at the University of Michigan I have had the privilege to “reboot” my research career at Sanford Burnham Prebys where I have had the opportunity to develop novel methodologies to understand cardiomyopathy. I have also had the opportunity to work with NASA scientists to do experiments on the International Space Station.
Education
Postdoctoral Fellow, Stanford University, Palo Alto, CA, Neurochemistry Postdoctoral Fellow, University of Texas Medical School, Houston, TX, Neuroscience NIMH PhD, Wesleyan University, Middletown, CT, Neuroscience NIMH B.A., Lehigh University, Bethlehem, PA, Biology
Prestigious Runding Awards or Major Collaborative Grants
2015-2020: NIH R01 HL132241-01A1 – Using Drosophila genetics to identify molecular links between ion channel dysfunction and pathological cardiac remodeling. (PI) 2013-2018 NASA NRA #NNH12ZTT001N – The effects of microgravity on cardiac function, structure and gene expression using the Drosophila model. (Co-I)
Honor and Awards
2014: Space Florida International Space Station Research Competition Winner – Co-investigator – One of three Basic Research proposals selected for launch aboard SpaceX3 – Mission completed, live flies returned on May 18,2014 2001: Excellence in Teaching Award, University of Michigan 1997: Excellence in Teaching Award, University of Michigan 1986-1988: National Institute of Mental Health Fellowship 1983-1985: National Institute of Mental Health Fellowship 1981: Sigma Xi Research Award 1980 MBL Scholarship, Neural Systems and Behavior Course 1971-1975: National Merit Scholarship, Lehigh University
Board Appointments
2018-present: Board member American Society for Gravitational and Space Research
Anatomical Systems and Sites
Cardiovascular System, Heart
Research Models
Drosophila, Larval Zebrafish Heart, Zebrafish
Techniques and Technologies
Biophysiology, Cellular and Molecular Imaging, Fluorescence Microscopy, Gene Silencing, Genetics, In vivo Modeling, Ion Channels, Live Imaging, Microarrays, Microscopy and Imaging, Molecular Genetics, RNA Interference (RNAi), Semi-automated Optical Heartbeat Analysis (SOHA), Systems Biology, Transgenic Organisms
The Ocorr Lab is investigating the cellular and molecular basis of adult heart function and cardiomyopathies using the genetic model system Drosophila.
We use functional, electrophysiological, biochemical and immunohistochemical techniques that allow us to examine the roles of genes and gene products in cardiac channelopathies and stress-related cardiomyopathies.
Our lab pioneered the development of a novel methodology (Semi-automatic Optical Heartbeat Analysis, SOHA) that permits the quantification of heartbeat parameters in model systems with small hearts.
Using this system we have identified several ion channels in the fly heart that play prominent roles in repolarization of the human heart and cause arrhythmia in both the fly and in humans when mutated. We also have developed a number of other disease models including a diabetic-like cardiomyopathy induced by high sugar diet and hypoxia-induced cardiomyopathy.
Recently we have begun collaborations with NASA (by winning a Space Florida International Space Station Research Competition). We are using the fly to uncover the molecular/cellular basis for cardiac and muscle atrophy in astronauts exposed to extended periods of microgravity despite extensive exercise regimes aboard the ISS. Our flies were launched aboard SpaceX 3 for a month-long exposure to micro-gravity.
Karen Ocorr’s Research Report
My lab is working to understand the cellular and molecular basis of heart disease. One project is focused on the genetic basis of Atrial Fibrillation. This project is a collaborative one with the lab of Alexandre Colas. We are combining two model systems, the fly in my lab and human induced cardiomyocytes in his lab, to identify AFib genes that have been implicated from patient studies. Another project focuses on the role of metabolism in cardiomyopathies. This is because obesity and metabolic syndrome are linked to an increased risk of heart disease. We are studying the role of a key metabolic signaling molecule in hypertrophic cardiomyopathy. A separate effort is focused on the role of gravity in heart function. These studies will provide important information for future habitants of colonies on the moon and Mars. But they are also relevant to patients who are bedridden and to patients with muscle wasting (sarcopenia).
Timothy Huang completed his PhD at the University of Calgary (Canada) under Dr. Dallan Young, studying kinase pathways involved in mediating cell polarity in yeast. He studied mechanisms underlying actin cytoskeletal dysfunction in Alzheimer’s disease at Scripps with Dr. Gary Bokoch (La Jolla), before joining Dr. Huaxi Xu’s laboratory in 2012/2013.
Related Disease
Alzheimer’s Disease, Molecular Biology
Phenomena or Processes
Cell Biology, Cell Signaling, Neurobiology, Neurodegeneration, Neurogenesis, Neuron-Glia Interactions in Myelin, Proteolytic Pathways, Tyrosine Kinases
Anatomical Systems and Sites
Brain
Research Models
Human, Human Cell Lines, Mouse, Mouse Cell Lines
Techniques and Technologies
Biochemistry, Cellular and Molecular Imaging, Confocal Microscopy, Electrophysiology, Mass Spectrometry, Protein Engineering, Protein-Protein Interactions, Protein-Small Molecule Interactions, Proteomics
My research is focused on identifying and characterizing mechanisms of neurodegeneration in Alzheimer’s disease (AD) and other related neurodegenerative disorders, and identifying neuroprotective pathways that may be involved in slowing disease progression. Currently, my research is focused on the genetic AD risk factors SORLA (SORL1, LR11) and TREM2, which may be involved in attenuating pathogenic effects associated with cognitive decline. By implementing methods to enhance these neuroprotective pathways, we may be able to reverse neuronal and cognitive damage in AD, and possibly other associated disorders.
Timothy Huang’s Research Report
There are two projects that comprise my main focus: 1) SORLA: Sortilin-related receptor with LDLR class A repeats (SORLA, SORL1, or LR11) is a genetic risk factor associated with Alzheimer’s disease (AD). Although SORLA is known to regulate trafficking of the amyloid β (Aβ) precursor protein to decrease levels of proteotoxic Aβ oligomers, whether SORLA can counteract synaptic dysfunction induced by Aβ oligomers remains unclear. Our work indicates that SORLA interacts with the EphA4 receptor tyrosine kinase and attenuates ephrinA1 ligand–induced EphA4 clustering and activation to limit downstream effects of EphA4 signaling in neurons. Consistent with these findings, SORLA transgenic mice, compared with WT mice, exhibit decreased EphA4 activation and redistribution to postsynaptic densities, with milder deficits in long-term potentiation and memory induced by Aβ oligomers. Importantly, we detected elevated levels of active EphA4 in human AD brains, where EphA4 activation is inversely correlated with SORLA/EphA4 association. These results demonstrate a novel role for SORLA as a physiological and pathological EphA4 modulator, which attenuates synaptotoxic EphA4 activation and cognitive impairment associated with Aβ-induced neurodegeneration in AD. 2) TREM2: Although ligands for TREM2 such as ApoE have been previously identified, a definitive mechanism for TREM2 in AD has not been established. We recently determined that TREM2 directly binds Aβ oligomers with nanomolar affinity and R47H mutations attenuate TREM2/Aβ interaction. TREM2 deletion impairs Aβ turnover in primary microglia, and abrogates Aβ clearance in vivo. Aβ also triggers changes in microglial membrane potential which is impaired with TREM2 deletion. Moreover, TREM2 deletion attenuates Aβ-induced microglial morphogenic changes associated with activation, and inhibits Aβ-mediated induction of proinflammatory cytokine expression. Together, these results indicate that TREM2 may have opposing neuroprotective roles in mediating microglial Aβ clearance and turnover, while concurrently transducing potentially neurotoxic Aβ-induced inflammatory signals. To further define a role for TREM2 in Aβ clearance and Aβ-mediated microglial activation/cytokine expression, we plan to exploit use of TREM2 R47H knock-in and TREM2 WT and R47H overexpression mouse models currently housed in our laboratory to determine whether impaired TREM2/Aβ interactions can impair microglial response in the presence of Aβ. This provides essential groundwork in future strategies to optimize neuroprotective TREM2 Aβ clearance while limiting Aβ-induced microglial inflammation.
Sanford Burnham Prebys scientist publishes two papers that bring us one step closer to understanding—and potentially treating—the devastating condition. For
Dr. Blaho began her research career focused on how bioactive lipids contribute to the innate immune response against bacterial infection, characterizing roles for eicosanoids in the generation and resolution of Lyme arthritis pathology. The wild diversity of lipid species led Dr. Blaho to Weill Cornell Medical College in New York City to pursue postdoctoral training in the field of sphingolipids, particularly sphingosine 1-phosphate (S1P), and its receptors. Advancing to Instructor at Weill Cornell and later, Research Assistant Professor at Sanford Burnham Prebys, Dr. Blaho continued her research in lipid chaperones and receptor signaling, with an emphasis on cell-type differential effects on hematopoiesis and immunity in response to cell stressors. Dr. Blaho is now a Research Associate Professor in the Center for Neurologic Diseases.
“The immune system has the power to protect us from invading pathogens and cancer or to initiate a ‘self-destruct’ sequence that consumes us with inflammation and autoimmunity. It is fascinating to me that a simple ubiquitous fat molecule like S1P can control the birth and destiny of immune cells.”
Education
2014-2016: Instructor, Weill Cornell Medicine, Pathology and Laboratory Medicine and Neuroscience 2009-2014: Post-doctoral training, Weill Cornell Medicine, Pathology and Laboratory Medicine 2007-2009: Post-doctoral training, University of Missouri, Columbia, Veterinary Pathobiology 2007: PhD, University of Missouri, Columbia, Molecular Microbiology and Immunology – BS
Funding Awards and Collaborative Grants
National Heart, Lung, and Blood Institute R01 American Heart Association Scientist Development Grant 2014-15: Leon Levy Neuroscience Foundation Grant 2015: Foundation LeDucq SphingoNet Young Investigator Grant 2009-12: National Cancer Institute Individual Ruth L. Kirschstein Post-doctoral Fellowship
Honors and Recognition
2017: British Journal of Pharmacology Lecture: FASEB Summer Research Conference on Lysophospholipids and Related Mediators–from bench to clinic. 2014: Leon Levy Foundation Neuroscience Fellow 2010: Keystone Scholarship, Bioactive Lipids: Biochemistry and Diseases 2008: Keystone Scholarship, Eicosanoids and Other Mediators of Chronic Inflammation 2007: Young Investigator Award in Inflammation, Eicosanoid Research Foundation 2004: National Academy of Sciences Christine Mirzayan Policy Fellow, Institute of Medicine Board on Health Sciences Policy
Phenomena or Processes
Adaptive Immunity, Apoptosis and Cell Death, Cell Signaling, G-Protein Coupled Receptors, Hematopoiesis, Inflammation, Innate Immunity
Anatomical Systems and Sites
Hematopoietic System, Immune System and Inflammation, Vasculature
Research Models
Human Adult/Somatic Stem Cells, Mouse, Mouse Somatic Stem Cells, Primary Cells
Techniques and Technologies
Cell Biology, Cellular and Molecular Imaging, Confocal Microscopy, Fluorescence Microscopy, Gene Expression, Lipid Bilayers, Mass Spectrometry
The lipid sphingosine 1 phosphate (S1P) is found in high levels in the blood and lymph and is primarily carried by the protein ApoM, found on HDL. S1P can affect the cardiovascular, nervous, and immune systems via interaction with cell surface-expressed receptors, S1P1-5. My work is determining how changing S1P carrier or receptor expression and signaling can affect cells of the immune system, particularly in the bone marrow, and how this alters their ability to respond to stress or infection.