Alexander Strongin earned his PhD from Moscow State University in Russia in 1972 and his D.Sci. degree from the Institute of Microbial Genetics in Moscow in 1983. From 1982 to 1988, Dr. Strongin was head of the Laboratory of Functional Enzymology at the Institute of Genetics of Microorganisms in Moscow. He served as head of the Department of Biotechnology and Laboratory of Protein Engineering, Institute of Molecular Genetics, Russian Academy of Sciences, Moscow, from 1988 to 1990. From 1990 to 1994, he was a visiting professor of biochemistry in the Division of Dermatology at Washington University School of Medicine, St. Louis, Missouri. Dr. Strongin has worked in the La Jolla area since 1994, as senior staff scientist in the Biology Division at General Atomics, 1994-1995, and as senior staff scientist at the La Jolla Institute for Experimental Medicine, 1995-1999. Dr. Strongin joined Sanford Burnham Prebys on September 1, 1999.
Related Disease
Anthrax, Arthritis, Brain Cancer, Breast Cancer, Cancer, Hepatitis C, Multiple Sclerosis, Prostate Cancer, Type 1 Diabetes
Tumors produce unique enzymes, which by degrading the normal tissue provide an opportunity for tumor to grow in size and metastasize. These enzymes are called matrix metalloproteinases or MMPs. MMPs are a primary target for the design of anti-cancer pharmaceuticals. Our work aims to lay the foundation from which efficient therapeutics could ultimately be derived. Dr Strongin’s research is focused on the fundamental mechanisms involving MMPs in the processes of cell migration, cell proliferation and metastasis. The far-reaching goals of this pioneering research are to gain an understanding of how MMPs by cleaving the surrounding tissue and by affecting cell surface receptors govern locomotion of malignant cells. This knowledge is critical for design of efficient pharmaceuticals that may find applications in a variety of disease conditions including cancer, arthritis and stroke.
Alex Strongin’s Research Report
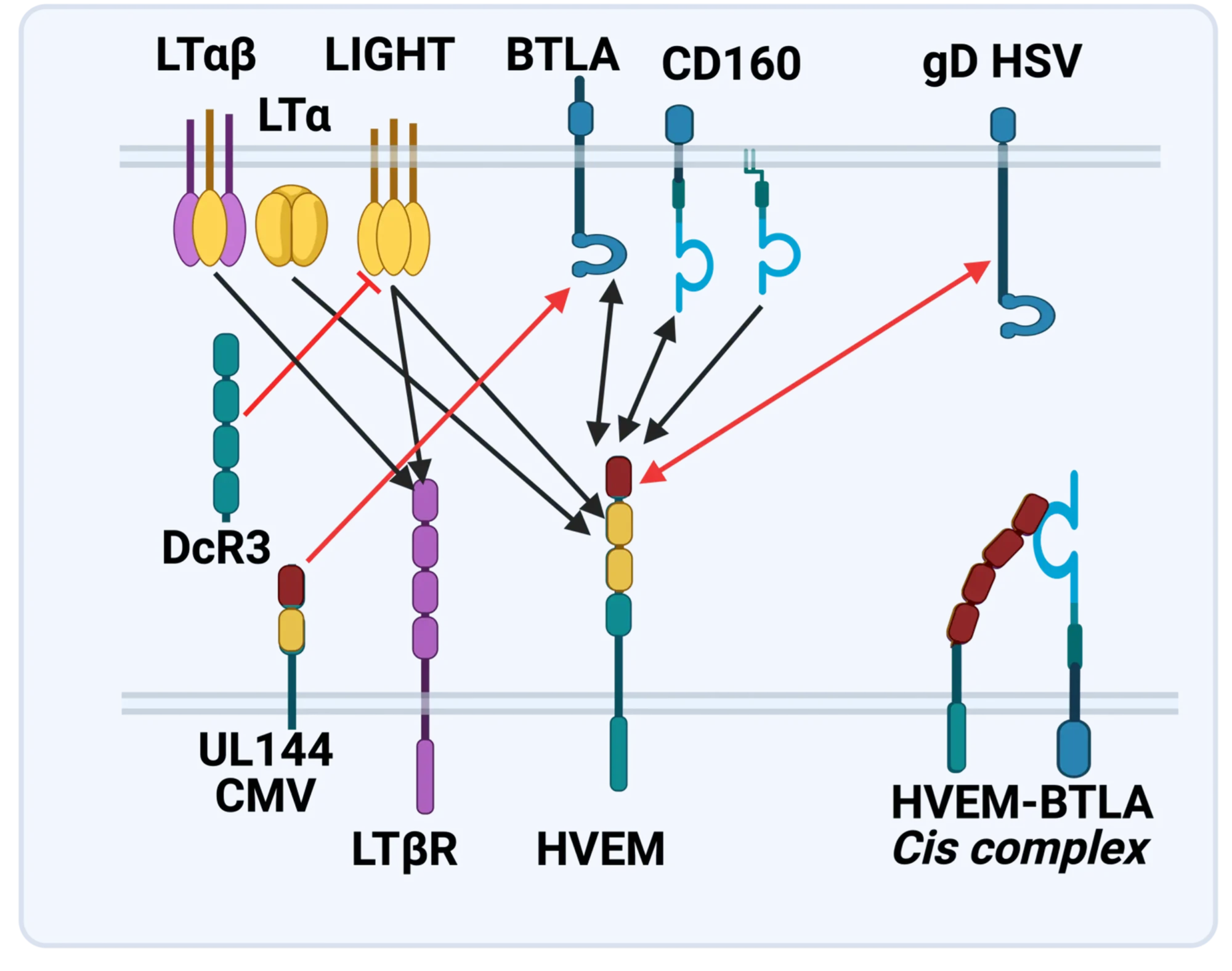
Degradation of the extracellular matrix and tissue remodeling play critical roles in tumor progression, particularly in invasion, metastasis, and neovascularization. Matrix metalloproteinases (MMPs) are essential for matrix proteolysis. Understanding the roles of MMPs in tumor growth and angiogenesis is of paramount importance to novel strategies for treatment of malignant tumors. Our primary goal is to characterize mechanisms of MMP activation involved in spatial and temporal control of focal proteolysis and to increase knowledge of cell-mediated matrix remodeling. Recently, we showed that heteromolecular assemblies involving MMPs and integrins that can simultaneously activate and dock MMPs are present on the surfaces of tumor cells. Apparently, these mechanisms of MMP activation are instrumental in clustering activated proteinases and integrins at the protrusions at the invasive front of migrating cells and at the invasive front of tumors (see Figure).
Interactions between MMPs and integrins, critical to spatial and temporal control of proteolysis, most likely are common for many cell types. To test our hypotheses and to outline novel approaches to control the activity of MMPs, we are examining mechanisms involved in activation, docking, and cross talk of MMPs and integrins in vivo and in vitro. We expect to experimentally show at the cell and protein sequence levels how the temporal and spatial regulation of matrix degradation is linked to dynamic changes of cell shape and cytoskeleton and to identify molecular mechanisms of cell motility. The immediate results of these studies will be improved diagnosis and treatment of malignant neoplasms.
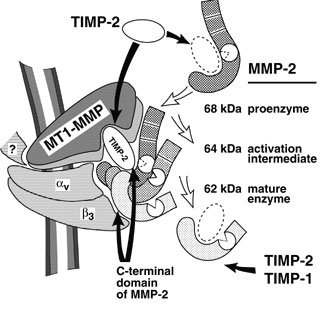
Schematic depiction of the MMP-integrin molecular assembly. Inhibitory effects of tissue inhibitors of metalloproteinases (TIMPs) and the C-terminal domain of MMP-2 are shown by black arrows. The conversions of MMP-2 are shown by open arrows. The question mark stands for a cytoskeletal protein involved in a complex with membrane type-1 matrix metalloproteinase (MT1-MMP). This assembly is critical for docking, activation, and cross talk of MMPs and integrins and is intimately involved in regulating focalized matrix degradation and cell locomotion. MT1-MMP is in immediate proximity to integrin alpha vbeta 3. Both MT1-MMP and the integrin associate with the cytoskeleton. TIMP-2 links MT1-MMP (“a receptor”) and the secretory MMP-2 proenzyme. The second molecule of MT1-MMP (“an activator” that is free of TIMP-2) activates integrin alpha vbeta 3 by limited proteolysis of the beta 3 integrin subunit. The activator initiates the activation of pro-MMP-2 by cleaving the N-terminal part of the 68-kDa MMP-2 latent zymogen. The 64-kDa activation intermediate of MMP-2 efficiently associates with activated integrin alpha vbeta 3 via the C-terminal domain of the enzyme. The 64-kDa to 62-kDa autocatalytic maturation occurs if MMP-2 is complexed with the integrin. The mature MMP-2 enzyme transiently associated with the integrin at the surface of tumor cells accelerates the directional invasion of the cells. Additionally, if transiently complexed with the integrin at the surface of host stromal cells, the enzyme facilitates tumor neovascularization. TIMPs, including TIMP-2 and TIMP-1, efficiently inhibit the activity of soluble MMP-2. By providing links between integrins, MMPs, and TIMPs and, more generally, between cell shape and focal proteolysis, this model represents basic mechanisms of pro-MMP-2 activation and illustrates a coordinated interplay of inhibitors, proteinases, and integrin adhesion receptors at cell surfaces.