This year’s event centered on CDG, a rare genetic condition that affects around 1,500 people worldwide.
With more than 270 attendees and 30 world-renowned scientists and clinicians, Sanford Burnham Prebys’ 11th Annual Rare Disease Day Symposium was officially the largest in the event’s history. This year’s discussion centered on congenital disorders of glycosylation, or CDG, a rare genetic condition that affects around 1,500 people worldwide.
Malin Burnham, T. Denny Sanford and Debra Turner, honorary trustees of Sanford Burnham Prebys, opened the three-day meeting with moving remarks. A theme emerged: Medical advances are accelerated when key stakeholders work together—including scientists, physicians, people with CDG and their families, patient advocacy groups, granting agencies, industry and philanthropists.
“Our annual symposium aims to create an ecosystem in which key stakeholders can collaborate and communicate,” said Hudson Freeze, PhD, director of the Human Genetics Program at Sanford Burnham Prebys and symposium chair. “Together, we celebrate our successes, focus on challenges and discuss the future of CDG research.”
Presenters at the meeting described the power of the ecosystem. Since the first conference a decade ago where scientists reported on the basic biology of CDGs, today we have promising clinical trials for the most common CDG mutation. In addition, a historic nationwide effort to establish the natural history of CDGs launched this year—which addresses decades of unresolved questions and helps remove barriers to starting clinical trials.
First discovered in the 1990s, scientists now know that CDG is caused by mutations that disrupt the body’s normal process of attaching sugars to proteins. Children with CDG have varying degrees of speech and language difficulty, poor balance, motor control, vision problems, hearing impairments and seizures.
Because CDG is rare, many patients bounce between doctors and clinics for years before they receive an accurate diagnosis. At the symposium, an innovative session called the “Doctor-is-in” session connects families with medical researchers and clinicians in small groups. For some medical researchers, the session is the first time they have met a person with CDG.
The conference was co-organized by Sanford Burnham Prebys and CDG CARE, a nonprofit organization founded by parents seeking information and support for CDG.
Institute News
Sanford Burnham Prebys scientist joins historic effort to help children with rare disease
Hudson Freeze, PhD, professor of Human Genetics at Sanford Burnham Prebys, has joined a historic effort that establishes—for the first time—a nationwide network of 10 regional academic centers, Sanford Burnham Prebys researchers and patient advocacy groups to address decades of unresolved questions surrounding congenital disorders of glycosylation, or CDG, a rare disease that affects children. The consortium is funded by a $5 million, five-year grant from the National Institutes of Health (NIH).
“We are extremely pleased that the NIH is investing in an initiative that will improve the lives of people affected by CDG,” says Freeze, who leads efforts to develop and validate disease biomarkers that will aid in diagnoses, and measuring treatment benefits during clinical trials. “Although globally the number of people living with CDG is relatively small, the impact on the lives of these individuals and their families can be profound. We look forward to working with the patients, families, physicians, scientists and other stakeholders focused on this important study.”
CDG is caused by genetic mutations that disrupt how the body’s sugar chains attach to proteins. First described in the 1990s, today scientists have discovered more than 140 types of mutations that lead to CDG. Symptoms are wide-ranging, but can include developmental delays, movement problems and impaired organ function. Some children may benefit from a sugar-based therapy; however, developing treatments for those who need alternative treatment options has been hindered by a lack of natural history data—tracking the course of the condition over time—comprehensive patient registry, and reliable methods to establish the CDG type.
Working together, the consortium will overcome these hurdles by:
Defining the natural history of CDG through a patient study, validating patient-reported outcomes and sharing CDG knowledge
Developing and validating new biochemical diagnostic techniques and therapeutic biomarkers to use in clinical trials
Evaluating whether dietary treatments restore glycosylation to improve clinical symptoms and quality of life
Freeze will lead the efforts to develop and validate biomarkers for CDG, working alongside the Children’s Hospital of Philadelphia and the Mayo Clinic. The principal investigator of the CDG Consortium Project is Eva Morava, MD, PhD, professor of Medical Genetics at the Mayo Clinic. The patient advocacy groups involved are CDG CARE and NGLY1.org.
Sanford Burnham Prebys and CDG Care will host the 2020 Rare Disease Day Symposium and CDG Family Conference from February 28 to March 1 in San Diego, which welcomes researchers, clinicians, children with CDG and their families, and additional CDG community members. Register to attend.
Institute News
Parents gain answers about their child’s mysterious condition, thanks to SBP scientists
For the parents of a six-year-old Hispanic boy and a seven-year-old Qatari girl, answers remained elusive. Both children had alarming symptoms, including developmental delays, uncontrollable seizures and “floppy baby syndrome” (hypotonia). But despite doctors’ best efforts, the origin of the disease remained unknown.
Now, these two children are linked by rare mutations in a gene called FUK—providing their families and doctors a better understanding of the cause of their medical conditions. Using biochemical techniques to analyze the boy’s cells, Sanford Burnham Prebys Medical Research Institute (SBP) scientists determined that a malfunctioning enzyme called fucokinase is to blame—caused by a mutation in the FUK gene. Because cells from the girl weren’t available, computer modeling was used—and indicated this same mutation likely caused the disease. The study published in the American Journal of Human Genetics.
Like a molecular spark plug, the fucokinase enzyme ignites one step in a cellular communication cascade—which culminates in the linkage of a sugar, fucose, to another carbohydrate. This final fucose-carbohydrate product is important for immune system regulation, tissue development, cell adhesion (“stickiness” to the environment) and more.
Based on these findings, the scientists now know the condition is a congenital disorder of glycosylation (CDG), an umbrella term for disorders caused by abnormal linking of sugars to cellular building blocks, including proteins, fats (lipids) and carbohydrates. Although more than 130 types of CDGs exist, the boy and girl are the only known living individuals who have this mutation.
“Our hope is that by reporting this information, we will help doctors grant more answers to patients and their loved ones,” says Hudson Freeze, PhD, senior author of the paper and director and professor of the Human Genetics Program at SBP. “Based on our findings, genetic databases around the world will now note this mutation causes disease—a potentially life-changing shortcut in the quest for answers.”
The researchers analyzed skin and immune cells that were collected from the boy. They observed reductions in the amount of the fucokinase enzyme—as much as 80 percent in skin cells and more than half in immune cells, compared to a control protein. Consistent with these findings, downstream products typically created by fucokinase weren’t incorporated into the final fucose-carbohydrate product—indicating the enzyme was not working.
Because cells from the girl were not available, the scientists used computer modeling to predict the impact of her FUK gene mutation. This approach indicated the mutation occurs at an important site on the enzyme that would likely cause disease.
“We know that dampening down the activity of the FUK gene is linked to metastatic cancer—a deadly event that occurs when tumors gain the ability to travel throughout the body,” says Freeze. “In addition to providing long-awaited answers to these families, these findings could help us understand how certain cancers spread throughout the body, including liver, colorectal and skin cancers (melanoma).”
Both children were identified through the National Institutes of Health’s Undiagnosed Diseases Network, which is designed to accelerate discovery and innovation in the way patients with previously undiagnosed diseases are diagnosed and treated.
Additional study authors include: Jill Rosenfeld, Lisa Emrick, MD, Lindsay Burrage, MD, PhD, Brendan Lee, MD, PhD, William Craigen, MD, PhD, Baylor College of Medicine; Mahim Jain, MD, PhD, Johns Hopkins School of Medicine; David Bearden, MD, University of Rochester School of Medicine; and Brett Graham, MD, PhD, Baylor College of Medicine and Indiana University School of Medicine. The study’s DOI is https://doi.org/10.1016/j.ajhg.2018.10.021
Research reported in this story was supported by National Institutes of Health (NIH) grants R01DK099551, U01HG007709, and K08DK106453; Baylor College of Medicine Intellectual and Developmental Disabilities Research Center (U54 HD083092), Diana & Gabriel Wisdom and the Rocket Fund. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Interested in keeping up with SBP’s latest discoveries, upcoming events and more? Subscribe to our monthly newsletter, Discoveries.
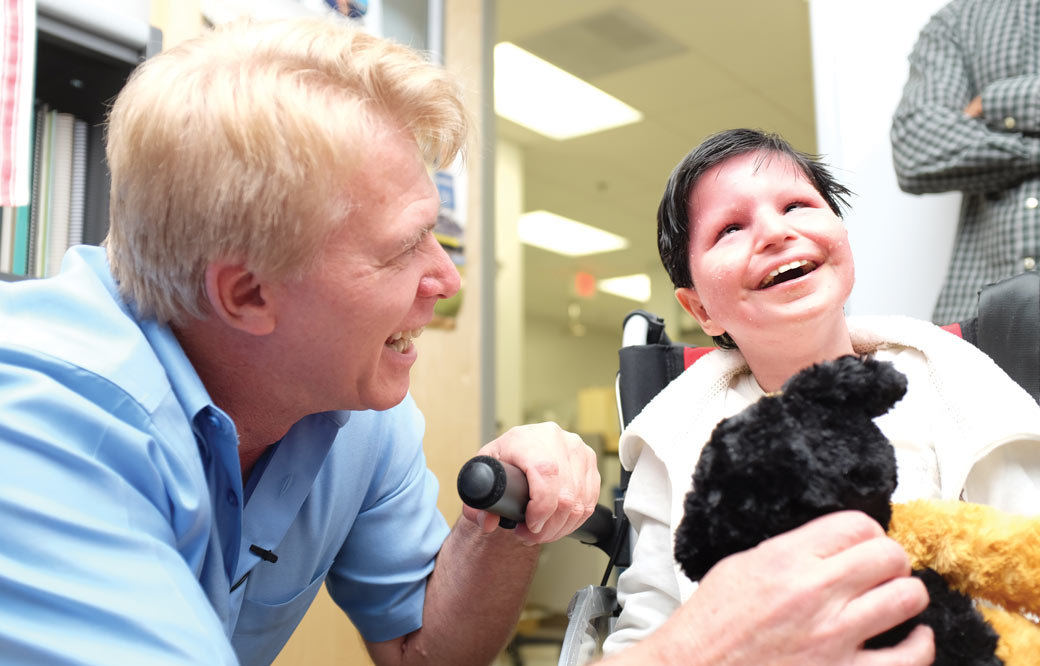
“I’ve got really cool stuff in my body,” 6-year-old Baraa Ismail proclaimed to Hudson Freeze, PhD, professor in the Human Genetics program at Sanford Burnham Prebys Medical Discovery Institute (SBP).
And, indeed, he does.
Baraa and his mother, Sara, didn’t know it at the time, but he was born with a rare change in his DNA that interfered with his body’s ability to attach a sugar to proteins—altering the course of his life.
From birth, Baraa struggled with eating. He dealt with upset stomachs and lethargy, which is unusual for a young child. Sara searched high and low for an answer, but doctor visit after doctor visit, year after year, Baraa remained undiagnosed.
After four years of uncertainty, Sara connected with Dr. Tawhida Yassin Abdel Ghaffar. She suspected a rare condition called congenital disorders of glycosylation (CDG) and ordered a test. Her instincts were correct—Baraa had one form of CDG. More than 130 types of the condition exist.
In addition to working with her doctor, Sara was introduced to a new online community of parents and individuals with CDG. It was through a private Facebook group that she connected with another parent whose child has CDG. He told her, “You have to talk to Hudson Freeze at SBP.”
For more than three decades, Freeze and his team have studied CDG with the ultimate goal of developing a treatment. When Freeze heard from Sara, he recommended that she talk to her doctor about giving Baraa mannose, a nutritional supplement.
Years ago Freeze helped discover that mannose can treat one specific form of CDG—incredibly reversing symptoms as quickly as days sometimes (note: Freeze is not a medical doctor). But it only works if a person has one kind of mutation; treatments are still limited for the 129 other types of CDG that exist. Freeze reasoned that even if he didn’t have Baraa’s genetic sequence in hand, if the boy had that mutation, the mannose would work.
Incredibly, it did. Within a month, Baraa’s energy was back. And today, Baraa is a Flash-loving, book-devouring little boy who loves to run and sing.
Baraa was doing so well that Sara even decided to take a vacation for the first time in years. She and Baraa traveled from their home in Egypt to visit her brother in Irvine, California—which happened to be a short drive from SBP. Sara reached out to Freeze, whom she calls “the man who saved my son’s life,” to see if a visit was possible. Freeze was delighted to meet with her and Baraa and give them a tour of his lab.
“Really, my role was very small in this story,” says Freeze. “But what this illustrates is the importance information has for these families. A simple piece of information changed someone’s life. We’d love to grow so we can eventually become a true hub of information for these families—and help even more people like Sara and Baraa.”
A father’s love never dies, even when his child has succumbed to a fatal childhood disease. Alex Syed paid tribute to his daughter, Aria, by running a marathon through the majestic Grand Canyon in May 2018. Ten years ago, Aria passed away after suffering from congenital disorders of glycosylation, known as CDG—a disease that affects the way proteins and sugar molecules interact in the body.
In honor of his daughter, Syed raised more than $4,300 from 71 donors to fund important research in the laboratory of Hudson Freeze, PhD, professor and director of the Human Genetics Program at Sanford Burnham Prebys Medical Discovery Institute (SBP). Freeze is one of the world’s leading experts on CDG.
“Funding is always in short supply, so the donations that Alex raised in the name of Aria will bring us closer to finding a cure for other kids suffering from this devastating disease,” says Freeze. “Aria’s cells are in our lab to support experiments that will help build a database for possible therapies,” he adds.
To prepare for the run, which Syed dubbed “A Grand Canyon Aria,” he rose in the middle of the night to begin his 26-mile run from the South Rim via Bright Angel to North Rim, taking him over the mighty Colorado River.
Running through the Grand Canyon to raise money for Dr. Freeze’s research was Syed’s idea. He knows that rare genetic diseases do not receive a lot of funding from large pharma companies. “The almost-angelic work that Dr. Hud Freeze and his team are performing at SBP is truly unique,” says Syed.
Children born with the rare genetic disorder known as CDG often live for years before they receive a diagnosis. CDG—which stands for congenital disorders of glycosylation—can cause serious, sometimes fatal, malfunction of different organs and systems in the body, including the nervous system, muscles and intestines. Children with CDG have varying degrees of speech and language difficulty, poor balance, motor control, vision problems, hearing impairments and seizures.
CDGs are difficult to diagnose partly because there are only about 1,800 known cases worldwide. But through global networking and the unwavering determination of researchers and clinicians, new patients are being discovered every year, providing important information to parents to help them better understand what they are dealing with.
Hudson Freeze, PhD, director and professor of the Human Genetics Program at SBP, is a one of the scientific leaders helping diagnose new cases of CDG. Freeze and his colleague, Bobby Ng, recently led an international team charged with diagnosing three unrelated individuals thought to have a new type of CDG—but not confirmed. The work, published in the American Journal of Human Genetics, confirmed that the three had a specific kind of CDG never seen before, adding to the more than 125 existing types of CDGs.
“All CDG disorders are caused by mutations that impair glycosylation—the complex process by which cells build long sugar chains that are attached to proteins called glycoproteins,” explains Freeze. “These sugar chains are crucial for cellular growth, communication and essential cell functions.
“There are many genes involved in proper glycosylation,” says Freeze. “When two parents happen to carry a mutation on the same gene, they end up with a one-in-four chance of passing both mutated copies on their child, and that causes the disorder.”
Increasingly, babies and children with unexplained health problems such as developmental delays and organ dysfunction undergo whole-exome sequencing, a technique that sequences the part of the genome that encodes proteins.
“Exome sequencing is used to find mutations in genes, but sometimes we don’t know if the mutations found actually translate to a genetic condition like CDG,” says Ng.
“Our lab steps in when a suspected mutation is found in one of the many enzymes involved in glycosylation,” says Ng. “We perform biochemical tests to confirm that the mutation impairs the glycosylation process, helping families narrow in on a CDG diagnosis.”
“The three patients in the current study are the only confirmed cases of the FUT8-CDG type in the world,” says Freeze. “These very rare diagnoses are only made possible when physicians, researchers and parents reach out across continents to families who’ve had nothing but questions.”
For the past 8 years, SBP has organized an annual symposium in San Diego where scientists, doctors, and families gather to discuss the latest in science and medicine, and meet other families coping with rare diseases.
For more information on the 2018 SBP Rare Disease Day Symposium and CDG Family Conference, click here.
From a farmhouse in rural Iowa, Crystal Vittetoe is fighting for her two babies afflicted with congenital disorders of glycosylation, known as CDG. She and her family have raised over $37,000 from a single fundraiser, and the donations keep coming in. “If we don’t fight for research, we are surrendering to CDG,” says Vittetoe.
“What Crystal has done for our research at the Institute is incredible. She’s raised enough money to pay for half a postdoc’s salary to do research for one year, and now we need to find the other half,” says Hudson Freeze, PhD, director of the Human Genetics Program at Sanford Burnham Prebys Medical Discovery Institute (SBP). “We have so many projects we start and want to complete. We need more hands on the projects. And if a family needs help, we don’t turn anybody away,” he says.
CDG is a collection of genetic diseases that causes mental and physical developmental issues, which leads to severe damage to multiple organs like the liver, heart and intestines.
The Vittetoes have two young children with CDG—two-year-old daughter Everlee (in the photo above) and one-year-old son Breckyn. Vittetoe drove from Iowa to SBP in La Jolla, Calif., for the annual Rare Disease Day Symposium at SBP. There, she met other families, scientists, doctors as well as Freeze to learn about the latest research and treatments that can help their kids cope with her illness. Worldwide, there are less than 1,500 known cases of CDG where children are born with the genetic disorders.
Vittetoe realized from the family’s visit to SBP that much more research was needed to figure out why CDG happens and how to lessen the her children’s suffering. She was inspired to raise money for the Rocket Fund, in honor of John Taylor (Rocket) Williams IV who would’ve turned 10 years old this year. Sadly, he passed away at the age of two.
In the past year, Everlee has been hospitalized six times. During one episode, she was having an hourly seizure for 24 hours with the last one enduring for 3.5 hours. “It’s so stressful, no matter if she’s having a stroke-like episode or just needs fluids,” says Vittetoe.
With the help of family and friends, Vittetoe held a dinner and silent auction at Lebowski’s Rock ‘N Bowl in her hometown of Washington, Iowa with a population of just over 7,000. The three-hour inaugural event raised a phenomenal amount of money that even surprised Vittetoe. “We were blown away,” she said.
The bar donated 15% of the tab and a friend, who’s also a singer, volunteered the entertainment. Over 300 people contributed to a free-will dinner donation for delicious pork loin from the family’s hog farm and scrumptious sides whipped up by the children’s grandmother.
Substantial seed donations, along with gifts from local businesses, raised an enormous amount of funds at the silent auction. The Vittetoes have been farming in Iowa for generations, and Crystal’s husband Jonathan approached the local seed dealers who all said “yes” to helping out the kids. And of course, neighborhood farmers came to support the Vittetoes who always need seed for their crops.
People contributed checks from $10 to $5,000, and every dollar counted. Other families with CDG children drove over six hours from as far away as Minnesota and Illinois to show their support.
The giving doesn’t just stop with the fundraiser hosted by the Vittetoe family. Recently Crystal’s grandfather passed away in Colorado and the family asked for memorial donations to the Rocket Fund.
Vittetoe says, “It’s your babies and if you don’t do something, you’re just waving the white flag. We’re not waving the white flag. We just want to do something for them.”
Note:
The next SBP Rare Disease Day Symposium will be held on February 24, 2017. The day-long event will focus on Alagille syndrome, a genetic disorder that causes liver damage due to abnormalities in the bile ducts, which carry waste from the liver to the gallbladder and small intestine. For more information, click here.
Photo credit: Drish Photography.
Institute News
Study triples the number of known cases of a rare disease
The Rare Disease Day symposium on February 26-27 featured many fascinating talks from experts on numerous aspects of congenital disorders of glycosylation (CDGs), from fundamental work on glycosylation pathways to animal models to diagnosis in the clinic. Following are summaries of each presentation:
Lawrence Tabak, D.D.S, PhD, deputy director of the NIH—After presenting his research on glycosylating enzymes in the 1980s, which helped lay the foundation for understanding the processes that are impaired in CDGs, Tabak discussed several initiatives by the NIH, including the Precision Medicine Initiative and efforts to increase reproducibility.
William Gahl, MD, PhD, director of the National Human Genome Research Institute (NHGRI)—Gahl highlighted several successes of the Undiagnosed Diseases Program. Most relevant to the field of CDGs was the discovery of the gene underlying a new type of CDG, in which an enzyme responsible for generating a necessary precursor for protein glycosylation (uridine diphosphate) is inactivated. This work also found that supplementation with uridine was an effective therapy.
Shengfang Jin, PhD, scientist at Agios Pharmaceuticals Inc.—Jin presented her work on a mouse model of PMM2-CDG, which is caused by mutations in the gene for phosphomannomutase 2. Her research has identified a promising biomarker for PMM2-CDG, which is one of the more common types of CDG.
Richard Steet, PhD, associate professor at the University of Georgia—Steet’s lab is developing a new method of identifying which proteins are glycosylated by particular enzymes, which is important for understanding how each CDG-associated mutation leads to disease.
Reid Gilmore, PhD, professor at University of Massachusetts Medical School—Gilmore gave a detailed view of how two CDG-associated mutations, in isoforms of the same component (STT3A and STT3B) of a major glycosylating enzyme, oligosaccharyltransferase, impair protein glycosylation.
Robert Haltiwanger, PhD, professor at the University of Georgia—In another presentation on fundamental glycobiology, Haltiwanger described the function of two enzymes in the same pathway (fucosylation) inactivated in certain CDGs. Mutations in these enzymes underlie Peters plus syndrome and a single case of an unnamed severe CDG, respectively.
Marjan Huizing, PhD, staff scientist at the NHGRI—Using a mouse model of GNE myopathy, a progressive muscle disease caused by mutations in an enzyme required for protein sialylation, Huizing’s lab identified a therapy, supplementation with the sugar ManNAc, which is now in phase 2 trials, and identified a key biomarker. The mouse model also suggested that sialylation problems may be associated with certain kidney diseases, which is now under investigation.
Raymond Wang, MD, clinical geneticist at CHOC Children’s Clinic—Wang told the story of how he and scientific collaborators diagnosed an unusual case that initially appeared to be a CDG because of abnormal glycosylation. The disease-causing mutation was finally identified to be in mitochondrial translation, highlighting the similarities between CDGs and mitochondrial diseases.
David Beeson, PhD, professor at the University of Oxford—Beeson described a subset of congenital myasthenias caused by mutations in glycosylating enzymes, which have distinct symptoms from other myasthenias. These mutations likely cause this disorder by selectively impairing processing of the receptor by which muscle cells receive signals from nerves—the nicotinic acetylcholine receptor.
Lance Wells, PhD, professor at the University of Georgia— Wells summarized his work on the molecular basis of dystroglycanopathies, a subgroup of muscular dystrophies that arise from defects in O-mannosylation enzymes. Most recently, his lab resolved the puzzle of how mutations in an enzyme involved in a different form of glycosylation could cause this disease—they showed that the enzyme’s function had been incorrectly assigned.
Taroh Kinoshita, PhD, professor at Osaka University—Kinoshita is an expert on the addition of sugar-based anchors to lipids (GPI anchors), which link many proteins to the cell surface. He presented some of the extensive work from his team on how mutations in GPI-synthesizing enzymes cause disease, including identification of a therapy, vitamin B6, for seizures in GPI deficiencies.
Eva Morava, MD, PhD, professor at Tulane University Medical Center and the University of Leuven—Morava described preliminary results of a clinical trial of galactose supplementation to treat PGM1-CDG, in which patients are deficient in phosphoglucomutase-1 (this also impairs glucose metabolism). In these patients, galactose improves liver function and endocrine abnormalities and normalizes clotting factors.
Lynne Wolfe, MS, C.N.R.P.clinical research coordinator at the NHGRI—Wolfe discussed the CDG natural history study underway at the NIH—its goals and progress so far. The findings of this study will serve as a resource both for future diagnoses and for researchers in the field to correlate pathways with symptoms.
Tadashi Suzuki, D.Sci., team leader at the RIKEN Global Research Cluster—NGLY1 is different from other CDG-associated genes—it encodes a deglycosylating enzyme, which helps degrade glycosylated proteins that aren’t properly folded. Suzuki’s team has shown that inhibiting another deglycosylating enzyme, ENGase, prevents the formation of aggregates of misfolded proteins, suggesting that it could be a therapeutic target.
Hamed Jafar-Nejad, MD, associate professor at Baylor College of Medicine—Using fruit flies as a model, Jafar-Nejad’s lab is investigating how NGLY1 deficiency affects development. These flies replicate many of the features of human disease, including growth delay and impaired movement, so they could yield important insights into pathogenesis.
There are more than 7,000 rare diseases, but congenital disorders of glycosylation (CDGs) are among the cruelest. One particular condition, called ALG1, can have dire consequences. Affected children face intellectual disabilities, seizures, skeletal issues, facial deformities and many other problems.
“These are really sick kids,” says Hudson Freeze, PhD, professor and director of the Human Genetics Program at SBP. “Almost 45 percent die in the first several years, and many of these children will have severe developmental delays.”
Glycosylation is a critical biological process, in which sugar molecules are added to proteins to make them function properly. A protein that’s improperly glycosylated is like a car without a steering wheel – it simply can’t perform its job.
To make matters worse, correctly diagnosing ALG1 and other CDGs can be a long, stressful and expensive process. Sometimes families must wait months or years to find out what’s causing their child’s condition. And while genomic sequencing is beginning to make a difference, more must be done to diagnose sick kids and help parents make informed decisions.
One potential solution is disease markers – biochemical signatures that identify particular conditions. Armed with this information, clinicians could accelerate the diagnostic process with a simple blood test.
A Unique Sugar Molecule
Researchers may have found a marker for ALG1 and possibly other CDGs. In a paper published in the journal Clinical Chemistry, the team describes a unique sugar molecule that is particularly common in children with ALG1.
The sugar, a type of N-tetrasaccharide, was discovered by Miao He, PhD, who co-directs the Metabolic Disease Laboratory at The Children’s Hospital of Philadelphia. However, he had only a few patients and she was unclear on the molecule’s origin. Working closely with Freeze’s lab, she started hunting for the aberrant sugar in Freeze’s large collection of proven ALG1 patients.
“We looked at a number of kids with ALG1 and kept finding this abnormal sugar,” says Freeze. “It’s a sugar chain that doesn’t normally exist in nature. You can perform a very simple test, that costs just a few hundred dollars, and if you see this abnormality, you could get genetic confirmation and turn it around quickly.”
The beauty of this marker is that it narrows the field for genomic analysis. Rather than looking at a patient’s entire genome – billions of base pairs and more than 20,000 genes – clinicians can focus on the gene that may be causing the disorder, dramatically accelerating the diagnostic process.
Quickly diagnosing a rare disorder can help get kids into treatment, if treatments are available. But it can also help parents navigate the family planning process and inform prenatal testing. In the big picture, disease markers could be a critical adjunct for genomic testing.
“Genome and exome sequencing is the future, but it will require some biochemical confirmation to support the genomic test,” notes Freeze. “This marker can really help us shortcut the long diagnostic odyssey many parents must go through.”