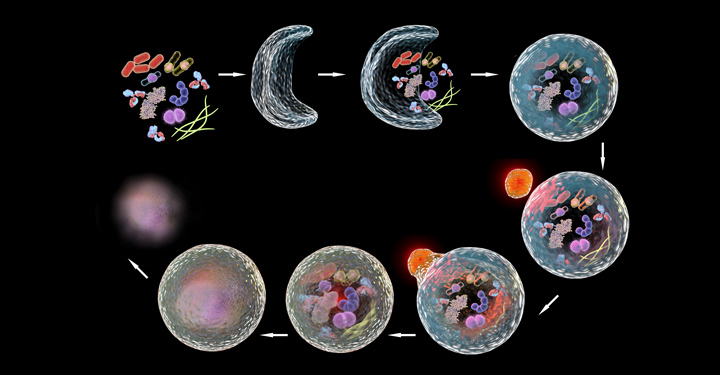
Autophagy genes help extrude protein aggregates from neurons in the nematode C. elegans.
Autophagy, which declines with age, may hold more mysteries than researchers previously suspected. In the January 4 issue of Nature Aging, it was noted that scientists from the Buck Institute, Sanford Burnham Prebys and Rutgers University have uncovered possible novel functions for various autophagy genes, which may control different forms of disposal including misfolded proteins—and ultimately affect aging.
“While this is very basic research, this work is a reminder that it is critical for us to understand whether we have the whole story about the different genes that have been related to aging or age-related diseases,” said Professor Malene Hansen, PhD, Buck’s chief scientific officer, who is also the study’s co-senior author. “If the mechanism we found is conserved in other organisms, we speculate that it may play a broader role in aging than has been previously appreciated and may provide a method to improve life span.”
These new observations provide another perspective to what was traditionally thought to be occurring during autophagy.
Autophagy is a cellular “housekeeping” process that promotes health by recycling or disposing of damaged DNA and RNA and other cellular components in a multi-step degradative process. It has been shown to be a key player in preventing aging and diseases of aging, including cancer, cardiovascular disease, diabetes and neurodegeneration. Notably, research has shown that autophagy genes are responsible for prolonged life span in a variety of long-lived organisms.
The classical explanation of how autophagy works is that the cellular “garbage” to be dealt with is sequestered in a membrane-surrounded vesicle, and ultimately delivered to lysosomes for degradation. However, Hansen, who has studied the role of autophagy in aging for most of her career, was intrigued by an accumulation of evidence that indicated that this was not the only process in which autophagy genes can function.
“There had been this growing notion over the last few years that genes in the early steps of autophagy were ‘moonlighting’ in processes outside of this classical lysosomal degradation,” she said. “Additionally, while it is known that multiple autophagy genes are required for increased life span, the tissue-specific roles of specific autophagy genes are not well defined.”
To comprehensively investigate the role that autophagy genes play in neurons—a key cell type for neurodegenerative diseases—the team analyzed Caenorhabditis elegans, a tiny worm that is frequently used to model the genetics of aging and which has a very well-studied nervous system. The researchers specifically inhibited autophagy genes functioning at each step of the process in the neurons of the animals, and found that neuronal inhibition of early-acting, but not late-acting, autophagy genes, extended life span.
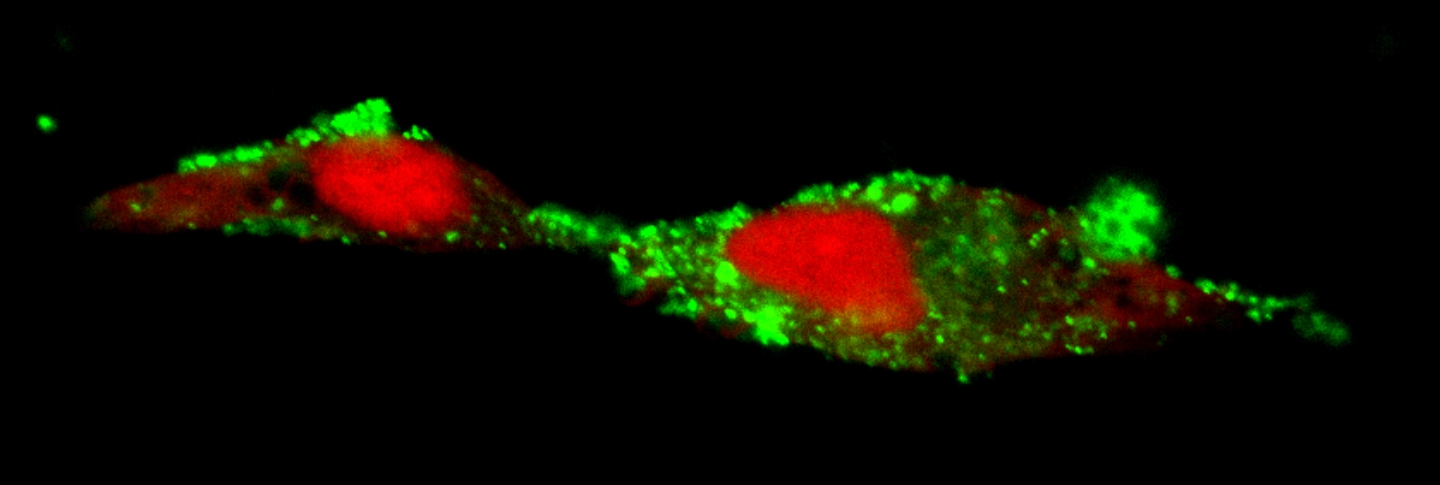
An unexpected aspect was that this life span extension was accompanied by a reduction in aggregated protein in the neurons (an increase is associated with Huntington’s disease, for example), and an increase in the formation of so-called exophers. These giant vesicles extruded from neurons were identified in 2017 by Monica Driscoll, PhD, a collaborator and professor at Rutgers University.
“Exophers are thought to be essentially another cellular garbage disposal method, a mega-bag of trash,” said Caroline Kumsta, PhD, co-senior author and assistant professor at Sanford Burnham Prebys “When there is either too much trash accumulating in neurons, or when the normal ‘in-house’ garbage disposal system is broken, the cellular waste is then being thrown out in these exophers.
“Interestingly, worms that formed exophers had reduced protein aggregation and lived significantly longer. This finding suggests a link between this process of this massive disposal event to overall health,” said Kumsta. The team found that this process was dependent on a protein called ATG-16.2.
The study identified several new functions for the autophagy protein ATG-16.2, including in exopher formation and life span determination, which led the team to speculate that this protein plays a nontraditional and unexpected role in the aging process. If this same mechanism is operating in other organisms, it may provide a method of manipulating autophagy genes to improve neuronal health and increase life span.
“But first we have to learn more—especially how ATG-16.2 is regulated and whether it is relevant in a broader sense, in other tissues and other species,” Hansen said. The Hansen and Kumsta teams are planning on following up with a number of longevity models, including nematodes, mammalian cell cultures, human blood and mice.
“Learning if there are multiple functions around autophagy genes like ATG-16.2 is going to be super important in developing potential therapies,” Kumsta said. “It is currently very basic biology, but that is where we are in terms of knowing what those genes do.”
The traditional explanation that aging and autophagy are linked because of lysosomal degradation may need to expand to include additional pathways, which would have to be targeted differently to address the diseases and the problems that are associated with that. “It will be important to know either way,” Hansen said. “The implications of such additional functions may hold a potential paradigm shift.”
DOI: 10.1038/s43587-023-00548-1