
Yu Yamaguchi earned his MD from Tohoku University in Japan in 1981, followed by a PhD in 1985, and training in obstetrics and gynecology at the same institute. Dr. Yamaguchi came to Sanford Burnham Prebys for his postdoctoral training. He was appointed to the staff in 1991.
Honors and Recognition
The Humanitarian Scientific Achievement Award, The MHE Research Foundation
The Kushima Prize, The Alumni Association, Tohoku University School of Medicine
Other Affiliations
Member, Scientific and Medical Advisory Board, The MHE Research Foundation
Related Disease
Alzheimer’s Disease, Arthritis, Autism Spectrum Disorders, Bone Mineralization Disorders, Epilepsy, Multiple Hereditary Exostoses
The goal of research in the Yamaguchi laboratory is to understand the role of proteoglycans and glycosaminoglycans in the context of development and human disorders. The general strategy is to define the role of proteoglycans and glycosaminoglycans by characterizing the phenotype of mutant mice lacking the synthesis of individual glycosaminoglycans. Specifically, mutant mice lacking the Ext1 and Has genes have been created to study heparan sulfate and hyaluronan, respectively. Recent progress in genetic studies in humans and mice has begun to reveal that deficiencies in glycosaminoglycans can be the causes and/or confounding factors of human childhood disorders. The Yamaguchi lab is now working to clarify the molecular mechanisms of two such disorders (multiple hereditary exostoses and autism) in order to develop new medical treatments.
For more information on the impact of Dr. Yamaguchi’s work, read letters from the patients with MHE.
Yu Yamaguchi’s Research Report
What Are Proteoglycans and Glycosaminoglycans?
Proteoglycans are a family of glycoproteins consisting of a core protein and a various number of long sugar chains called glycosaminoglycans attached to the core protein (Fig. 1). There are four classes of glycosaminoglycans; heparan sulfate, chondroitin sulfate, keratan sulfate, and hyaluronan (hyaluronic acid). Heparin, the anticoagulant widely used in clinics, is a specialized form of heparan sulfate. Although there is ample circumstantial evidence that these glycosaminoglycans have important biological functions, a complete understanding of their function and their relevance to human diseases requires genetic animal models.
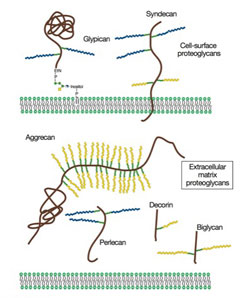
Figure 1. Proteoglycans consist of a protein core and one or more covalently attached glycosaminoglycan chains. From Esko, JD, Kimata, K., and Lindahl, U. Proteoglycans and Sulfated Glycosaminoglycans, In: Essentials of Glycobiology, CSH Press.
Developmental Roles Of Glycosaminoglycans
HEPARAN SULFATE – The Ext1 gene encodes an enzyme essential for the elongation of nascent heparan sulfate chains. As a result of genetic ablation of Ext1 using a ‘conditional knockout’ approach, heparan sulfate is eliminated from specific tissues and cell types. Our previous studies using brain-specific Ext1 conditional knockout have demonstrated critical roles of heparan sulfate in brain patterning, neurogenesis in the cerebral cortex, and pathfinding of various axon tracts (1)(2)(3)(4). More recently, conditional Ext1 knockout in developing limb bones revealed critical roles of heparan sulfate in the growth and patterning of bones and joint formation (5).
Moreover, our conditional Ext1 mutant mouse model has been distributed to more than 20 laboratories worldwide to characterize the role of heparan sulfate in various tissues and cell types, such as colon (6), kidney (7), lymphocytes (8), blood vessels (9), eyes (10), embryonic stem cells (11), and so on.
HYALURONAN – Three Has genes (Has1, Has2, Has3) encode the entire repertoire of hyaluronan synthases in mammalian cells. Genetic ablation of these genes, singly or in combination, results in a reduction or total elimination of hyaluronan, depending on the repertoire of Has expression in the given tissue. We created a conditional null allele of the Has2 gene, which is the predominant Has in many tissues. Our conditional Has2 knockout study targeted to the limb bud mesenchyme has revealed that hyaluronan plays a critical role in the proliferation and maturation of chondrocytes in the developing limb skeleton (12). Like Ext1 mutant mice, these Has2 conditional knockout mice are being used in more than a dozen laboratories worldwide for studies on the role of hyaluronan in various tissue and cell types.
Deficiencies in glycosaminoglycan synthesis can be the causes of childhood disorders
Recent progress in genetic studies in humans and mice has begun to reveal that deficiencies in glycosaminoglycans can be the causes and/or confounding factors of human childhood disorders. The research focus of our lab is to elucidate the molecular mechanisms of such disorders and to develop new medical treatments. We are currently studying two such disorders.
MULTIPLE HEREDITARY EXOSTOSES – One of the major diseases studied in our lab is Multiple Hereditary Exostoses (MHE; also known as Hereditary Multiple Exostoses [HME] or Multiple Osteochondroma [MO]). MHE is caused by a mutation in Ext1 (see above) or its related gene, Ext2. As mentioned above, these genes encode an enzyme necessary to produce heparan sulfate. MHE occurs in children of 0-12 years old. Although no comprehensive survey has been conducted, it is estimated that there are several thousand individuals affected by MHE in the US, which makes MHE one of the more prevalent among ’rare diseases’. Dr. Yamaguchi is a member of the scientific advisory board of the MHE Research Foundation and has been working to promote collaborations between basic scientists, academic physicians, and patient advocates.
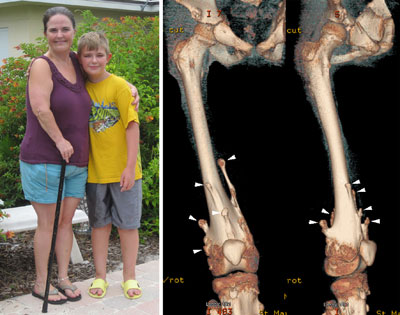
Figure 2. MHE patients, Carol and her 12-year-old son, Bruce. Shown on the right are three-dimensional CT images of Bruce’s right upper leg and knee joint area. Note that there are many bony protrusions (‘exostoses’), as indicated by white arrowheads. These tumors need to be surgically removed to prevent possible malignant transformation. Surgery is also needed to correct bone deformities and bone length inequalities. For example, Bruce and Carol have had 21 and 36 surgeries, respectively.
Children with MHE suffer from the formation of multiple –– sometimes as many as 100 –– bony tumors (osteochondromas) (Fig. 2). These bony tumors stunt their growth and can cause pain and disfigurement. Fortunately, the chance these tumors becomes cancerous is relatively low, partly because they are surgically removed as they develop. This means, however, children with MHE need to go through multiple surgeries over the course of their lives. There is currently no medical treatment for the disease.
Our lab is currently working to elucidate the molecular and cellular mechanisms of MHE. One of the major thrusts has been to create a mouse model that mimics the manifestations of human MHE. A long-term issue of MHE research has been the lack of mouse models that faithfully recapitulate the manifestation of human MHE; when Ext genes were inactivated in mice just as they are in human MHE patients, the mice failed to develop the symptoms of MHE.
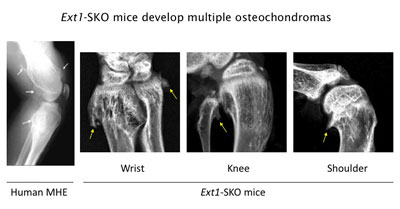
Figure 3. Ext1-SKO mice develop multiple osteochodromas in a pattern almost identical to human MHE. The X-ray images of the knee joint area of an MHE patient and the wrist, knee, and shoulder areas of Ext1-SKO mice. Osteochondromas are indicated by arrows. Ext1-SKO mice also mimic other skeletal deformities frequently seen human MHE, such as bowing of the forearm, the subluxation/dislocation of the radial head, and scoliosis (not shown).
Instead of knocking out the Ext1 gene in the whole mouse, we targeted the gene in only a small fraction of bone cells (Ext1-SKO mice). This minimalistic approach led to a mouse with all the physical manifestations of MHE, such as bony protrusions, short stature, and other skeletal deformities (Fig. 3)(13). The new mouse model answered some long-standing questions about MHE. Scientists had gone back and forth on whether osteochondromas observed in MHE are true tumors or just malformations of the bone. In this study, the tumors were made up of two cell types. A minority were mutant cells lacking Ext1, but, amazingly, most were normal bone cells. Hence, osteochondroma in MHE is not considered a true neoplasm in its strictest sense.
Our lab has been using this and additional mouse models to further dissect the pathogenic mechanism of MHE. Moreover, this mouse model provides new opportunities to test potential drugs to prevent osteochondroma formation and other clinical symptoms of MHE.
AUTISM – Children with MHE sometimes suffer from neurological and mental symptoms, which is not surprising because heparan sulfate is expressed and plays critical roles in the nervous system (1). The MHE community has long noticed the prevalence of autism-like behavioral traits in the patient population, and there are clinical reports describing the association of autism with MHE. Aided by the funds from the Sanford Health and the MHE Research Foundation, we have been studying the behavior of Ext1 mutant mice. Our preliminary data suggest that heparan sulfate has indeed a physiological function in the nervous system, and that its deficiency can cause behavioral deficits relevant to human autism. We are also analyzing DNA samples from individuals with autism for abnormalities in enzymes for heparan sulfate synthesis. Since heparan sulfate is a modulator of a number of neuronal molecules, we hope to identify functional networks of molecules underlying autism and other childhood mental disorders.
 Feb 1, 2022
Feb 1, 2022Sanford Burnham Prebys professor awarded $2.9 million to explore new answers to old questions in Alzheimer’s research
Feb 1, 2022Sanford Burnham Prebys professor Yu Yamaguchi, MD, PhD, has been awarded a $2.9 million grant from the National Institutes of Health
 Oct 21, 2021
Oct 21, 2021This enzyme is one of the hardest working proteins in the body
Oct 21, 2021Researchers have shown that a protein they identified plays a major role in the breakdown of hyaluronic acid, a compound…
 Mar 25, 2019
Mar 25, 2019Families find hope at our 10th Annual Rare Disease Day Symposium
Mar 25, 2019The unofficial theme of Sanford Burnham Prebys’ 10th annual Rare Disease Day symposium can be summarized in one word: hope.
 Jan 9, 2018
Jan 9, 2018Year in review: Top stories in 2017
Jan 9, 2018In the last 12 months, SBP scientists published 338 scientific papers—that’s almost a paper a day. We are proud of…
 Jul 14, 2017
Jul 14, 2017Genes and proteins go hand-in-hand
Jul 14, 2017Thanks to huge improvements in DNA sequencing technology, scientists have identified almost all the genes present in humans. Despite this…
 Mar 15, 2017
Mar 15, 2017Research may explain congenital deafness
Mar 15, 2017If you’ve heard of hyaluronic acid (HA), it’s probably as an ingredient in cosmetic products meant to help keep skin