Barbara Ranscht earned her PhD in Cell Biology/Developmental Neurobiology from the University of Tübingen, Germany in 1981. Her postdoctoral training was at King’s College in London, U.K., and the Massachusetts Institute of Technology in Cambridge, Massachusetts. Dr. Ranscht joined Sanford Burnham Prebys in 1987, and holds an adjunct professorship in the Department of Neurosciences at University of California, San Diego. From 1989 to 1992, Dr. Ranscht was the recipient of a McKnight Scholarship.
Education
1981: PhD, University of Tübingen, Germany Neurobiology
Related Disease
Attention-deficit hyperactivity disorder (ADHD), Cancer, Cardiovascular Diseases, Metabolic Syndrome, Multiple Sclerosis, Neurological and Psychiatric Disorders
Our lab studies cell surface interactions that regulate signaling networks in the nervous and cardiovascular systems and in cancer. With focus on the brain, we investigate membrane glycoproteins that enable crosstalk of neurons with the environment during circuitry development and disease. Our group has contributed seminal insights into the functions of Contactin-1 (Cntn1) and T-cadherin (Cdh13) in axon guidance, synapse formation and myelination using knock-out mouse models as well as biochemical, electrophysiological, histological and cellular approaches. Our current work explores the role of T-cadherin in brain circuitries enabling learning and memory formation, and determines how Contactin-1 functions in processes of myelination and remyelination. Exploiting our mouse genetic model, we uncovered a novel function for T-cadherin in protecting against stress-induced cardiac injury, and revealed disruptions of T-cadherin-mediated cellular interactions in cancer. In these latter contexts, our research identified T-cadherin as a major receptor for Adiponectin (Adipoq), a fat-secreted, circulating hormone that is of high clinical interest for its role in positively regulating energy balance and protecting against cellular insult. Current work in the lab aims to understand the molecular processes and signaling pathways regulated by T-cadherin or Contactin-1 in nervous system, cardiovascular and cancer functions with the goal to derive translational applications.
Barbara Ranscht’s Research Report
Functions of T-cadherin/Cdh13 in Neuronal Circuits Involved in Learning and Memory
Cell adhesion molecules play seminal roles in synapse formation, stability and function in the central nervous system (CNS). T-cadherin (Cadherin-13; Cdh13), a glycosylphosphatidylinositol-linked cadherin-type cell adhesion molecule discovered in our laboratory, is prominent in diverse structures of the CNS. Mutations in the human CDH13 gene are linked to neuropsychiatric disorders, most prominently Attention Deficit Hyperactivity Disorders and its comorbidities. Our lab generated mice deficient for T-cadherin gene expression (Tcad-KO) mice to uncover the T-cadherin-dependent neuronal circuitries and probe its contribution to cognitive function. Tcad-KO mice show no overt developmental, neurological or sensory deficiencies, and are indistinguishable from wild type littermates under normal housing conditions. However, challenging the mice in behavioral tests revealed defects in performing select learning tasks. Investigating hippocampal circuitry as a central relay station for learning and memory, we identified reductions in CA1 synaptic transmission and long-term potentiation along with reduced spine densities and maturation of hippocampal pyramidal neurons. Current work aims to link T-cadherin to specific synaptic circuitry, and probe its function through conditional T-cadherin ablation in select neuron populations of the interconnected circuitry to contribute principal understanding to human cognitive disorders.
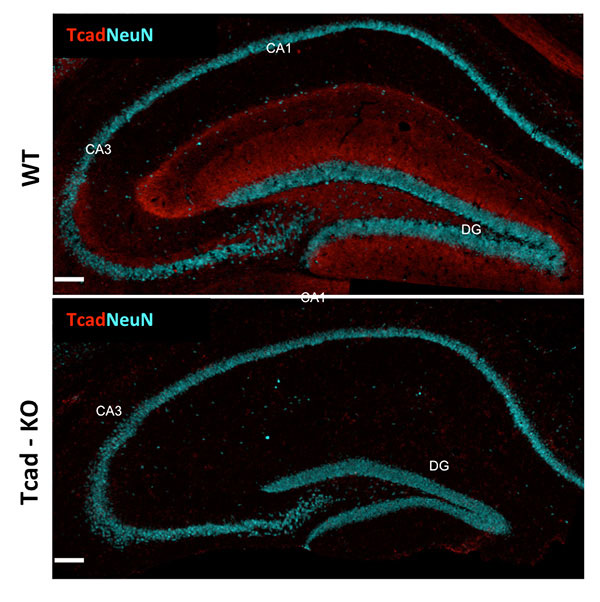
Figure 1: T-cadherin expression in adult hippocampus. T-cadherin staining delineates the dendritic layer of pyramidal neurons in CA1-CA3 subfields, and the molecular layer in the dentate gyrus. Staining is absent from the hippocampus of Tcad-KO mice. NeuN outlines the positions of pyramidal and granule neurons for orientation. Images by Hedie Badieh-Madhavi, PhD and Jinsha Liu, graduate student.
Mechanisms of Myelination and Remyelination
Myelin, a multilayered membrane sheath formed by oligodendrocytes around axons in the central nervous system (CNS) enables rapid nerve impulse conduction and sustains neuronal health and survival. Our lab is interested in the signals exchanged between axons oligodendrocytes during myelin formation and repair. Contactin-1, a GPI-linked cell surface glycoprotein, studied in our lab has emerged as one of the critical proteins to regulate axon-glia communication. In peripheral myelinated nerves, Contactin is solely expressed by neurons, and together with Caspr clusters at the paranodal axon domain to orchestrate formation of the septate-like axoglial junctions. In the CNS, Contactin delineates paranodes and nodes of Ranvier in mature myelin, and oligodendrocytes during the process of active myelination. We recently documented a vital dual role of Contactin-1 in central myelin formation: Contactin-mediated interactions between axons and oligodendrocytes regulate the extension and wrapping of myelin membranes, and loss of these interactions leads to hypomyelination in the mouse knockout model. The residual myelin is non-functional due to disrupted paranodal junctions that are characterized by loss of Contactin-associated paranodal Caspr, mislocalized potassium channels and disrupted transverse bands. Current work in the lab investigates the distinct contributions of Contactin in neurons and oligodendrocytes in mice with conditional Contactin-1 deletion, myelinating co-cultures and mouse models of myelin injury. If indicated, this work will form the basis for translational aspects of myelin repair.
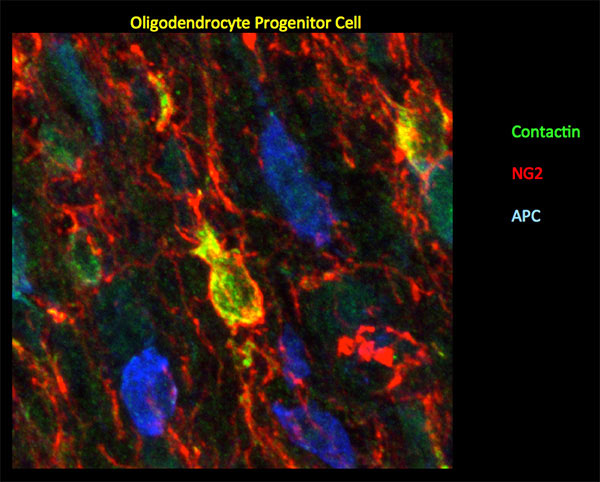
Figure 2: In mouse P10 optic nerve, Ccntactin (green) delineates oligodendrocyte progenitor cells demarcated by NG2 (red). Staining for Contactin does not overlap with APC (blue), a marker for mature oligodendrocytes. Image by Gulsen Colakoglu, PhD
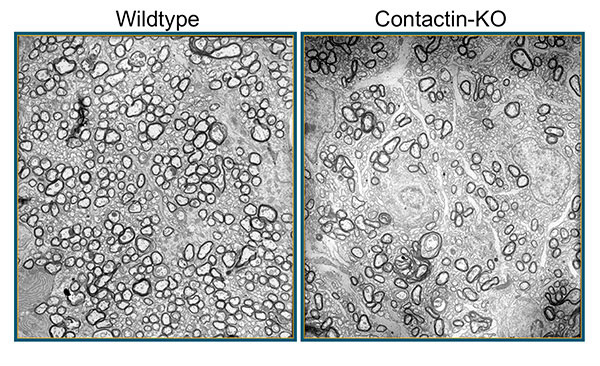
Figure 3: In mice deficient for Contactin-1, loss of axon-glia cross-talk leads to hypomyelination. Shown here are electron micropgraphs from P17 wildype and knockout (KO) optic nerves. Images Erik O. Berglund, MD, PhD
Adiponectin – T-cadherin/Cdh13 Signaling in Cardioprotection
Adiponectin is of significant clinical interest for its potency in counterbalancing the adverse effects of obesity-linked metabolic disease and cardiovascular dysfunctions. Research in the field is increasingly focusing on local actions of APN in its target tissues where binding to the cell surface leads to activation of intracellular signal transduction pathways that increase cellular energy and counteract adverse processes such as generation of reactive oxygen species, misbalanced cellular metabolism, and apoptotic cell death. Our lab identified T-cadherin as a major ligand-binding receptor for Adiponectin in the cardiovascular system (Denzel et al, 2010). T-cadherin effectively sequesters APN from serum and its absence leads to excessive APN concentrations in the circulation. Under cardiac stress, induced by pressure overload, T-cadherin is necessary to confer cardioprotection through APN. Downstream, APN engagement with T-cadherin activates the AMP-activated protein kinase (AMPK), a major downstream signaling target of APN. Since T-cadherin is a GPI-linked cell surface glycoprotein lacking the cytoplasmic region, it is our working model that T-cadherin connects through associated molecules to intracellular signaling pathways. We are exploring the heptahelical APN-receptors AdipR1/R2 as prime candidates while considering alternative possibilities.
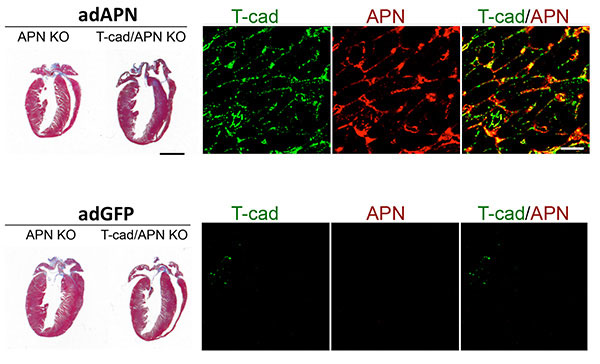
Figure 4: T-cadherin requirement in reverting exaggerated cardiac hypertrophy in APN-KO mice. Trichorme-stained hearts (left) show cardiac hypertrophy induced by pressure overload. Hypertrophy can be reverted in Adiponectin knockout (APN KO) mice by adenovirus-mediated expression of recombinant Adiponectin (adAPN; upper panel). In the absence of T-cadherin, APN-KO cardiac tissue does not respond (upper panel). Control virus expressing green fluorescent protein (adGFP) has no effect on the heart hypertrophic phenotype (lower panels). Mechanistically, as shown by immunohistochemistry, low T-cadherin expression in APN-KO hearts (Tcad, green, lower panel) is upreguated by adAPN (Tcad, green, upper panel) and binds adAPN which triggers downstream cardioprotective pathways. Data by Martin Denzel, PhD
Combining our cellular and functional analyses of T-cadherin with the extensive expertise in heart function at our Institute, we hope to make headway in determining APN-associated signaling pathways involving T-cadherin and AdipoR receptors. This work will fill the current knowledge gap of understanding APN interactions with its target cells in the cardiovascular system and may aid the development of novel molecular approaches for cardioprotection.
T-cadherin/Cdh13 in Cancer
T-cadherin has also emerged as one of the key molecules altered in diverse types of cancer. Epithelial-mesenchymal transition (EMT) during neoplasia is characterized by the down-regulation of T-cadherin/CDH13, and this silencing is linked to increased proliferative and invasive potential of human cancers. We used the mouse genetic model to investigate functions of T-cadherin in tumors. In the mouse mammary tumor virus-driven polyoma middle T (MMTV-PyV-mT) model, T-cadherin is down-regulated from neoplastic epithelial cells while it is upregulated in the tumor vasculature. Under conditions of T-cadherin deficiency, MMTV-PyV-mT induced tumor growth occurs with delayed onset and at reduced growth rates that result in prolonged animal survival over non-mutant mice. Our analyses identified reduced tumor vascularization as the major underlying mechanism for this beneficial outcome. T-cadherin’s pro-angiogenic role is mediated by the association of Adiponectin: The APN-deficient MMTV-PyV-mT tumor phenotype mirrors the defects in T-cadherin knockout mice, and in vitro silencing of T-cadherin in endothelial cells abrogates the cellular responses to APN. Our lab is now interested in understanding the mechanism by which T-cadherin binding to Adiponectin affects endothelial cell function and develop strategies to control tumor angiogenesis.
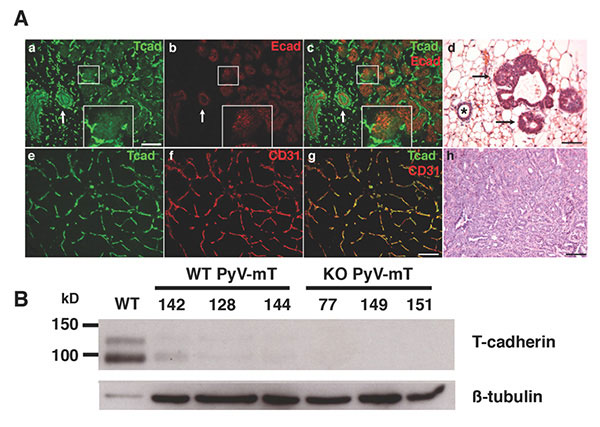
Figure 5: T-cadherin in the neoplastic mammary gland. A. In early stage MMTV-PyV-mT mouse tumors, T-cadherin (green) and E-cadherin (red) are down-regulated from developing neoplasias (a-c; central white box magnified as insert) while both cadherins are expressed on the non-cancerous ductal epithelium (white arrows in a-c), Histology shows the pathology of a normal duct (asterisk) and developing neoplasias (d, black arrows; scale bars 50 mm). In developed MMTV-PyV-mT tumors, T-cadherin (green in e) is upregulated on the CD31-positive tumor vasculature (f and g). The pathology of a MMTV-PyV-mT tumor is shown in f. B, T-cadherin is detected as a protein doublet of 95 kD and 115kD by Western blotting of normal mouse mammary fat pads and is down-regulated in the neoplastic mammary gland. Beta tubulin Is used as a gel loading control. No T-cadherin protein is detected in the T-cadherin-deficient (KO) MMTV-PyV-mT tumors. Data by Lionel Hebbard, PhD