Michiko N. Fukuda earned her PhD in biochemistry at the University of Tokyo in 1980. She did postdoctoral work at Fred Hutchinson Cancer Research Center in Seattle prior to her recruitment to Sanford-Burnham Medical Research Institute in 1982.
Education
1980: PhD, University of Tokyo, Biochemistry
1970: MS, University of Tokyo, Biochemistry
1968: BS, Tokyo University of Education, Botany
Related Disease
Breast Cancer, Cancer, Congenital Disorders of Glycosylation, Endometriosis, Glycosylation-Related Disorders, Inherited Disorders, Ovarian Cancer, Prostate Cancer, Testicular Cancer
Michiko Fukuda’s Research Report
Identification of Peptide that Delivers Drugs to Tumors
Chemotherapy effectiveness is often limited by drug toxicity in healthy tissues, although methods that spare normal cells by delivering drugs specifically to tumors may help to overcome this constraint. We have identified a promising tumor-targeted drug delivery vehicle known as the IF7 peptide. Using in vitro assays, we found that the IF7 peptide bound to the protein annexin 1 (Anxa1), which is known from previous studies by others to be enriched on the surface of tumor vasculature in several tumor types. When we injected a fluorescently labeled IF7 peptide into mice with tumors, fluorescent signals appeared in the tumors within one minute of injection. By contrast the tumors showed no fluorescence when mice were pre-injected with anti-Anxa1 antibodies that inhibits IF7-Anxa1 binding, suggesting that the peptide targets tumor by homing to Anxa1.
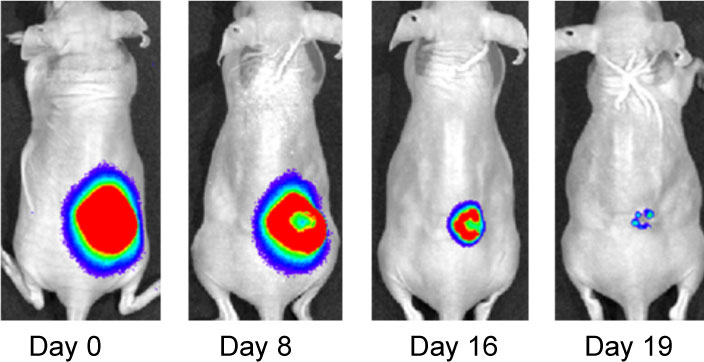
Effect of IF7-conjugated anti-cancer drug SN-38 on colon cancer mouse model. Activa cancer cells in a large tumor produce chemiluminescence which is captured by imaging. (Hatakeyama et al, 2011).
When IF7 peptide was conjugated with potent anti-cancer drug SN-38 and IF7-SN38 conjugate was injected intravenously to mice with tumors, IF7 could deliver SN-38 to tumors. We found that daily injections of an IF7-SN38 conjugate reduced a large tumor in the mouse without apparent side effects, whereas non-homing peptide-SN38 conjugate or with SN-38 alone did not reduce the tumors (Hatakeyama et al, 2011). The findings suggest that IF7 peptide may represent a clinically relevant vehicle for anti-cancer drugs.
Role of Trophinin in Human Embryo Implantation and Cancer
Invasion of the trophoblast into the endometrium, an essential element of embryo implantation, resembles invasion of malignant tumors. At the initial phase of implantation, the trophoblast and the uterine epithelium establish their first contact via their respective apical cell membranes. We have identified new molecules, trophinin, tastin, and bystin that mediate cell adhesion between trophoblastic cells and endometrial epithelial cells at the respective apical cell membranes. Trophinin is an intrinsic membrane protein, and tastin and bystin are cytoplasmic proteins. All of these molecules are strongly expressed in cells involved in embryo implantation in humans. However, trophinin is not expressed in human endometrial epithelia throughout the hormonal cycle, except only those cells located close to the implanting blastocyst. Trophinin expression by endometrial epithelia is induced by human chorionic gonadotrophin (hCG) derived from the implanting embryo (Sugihara et al, 2008). While embryos invade maternal cells (Sugihara et al, 2007), maternal tissue accepts embryos. We asked what happens in the maternal epithelia when trophinin-mediated adhesion takes place, and found that trophinin-mediated cell adhesion triggers an apoptotic signal in maternal epithelial cells (Tamura et al, 2011).
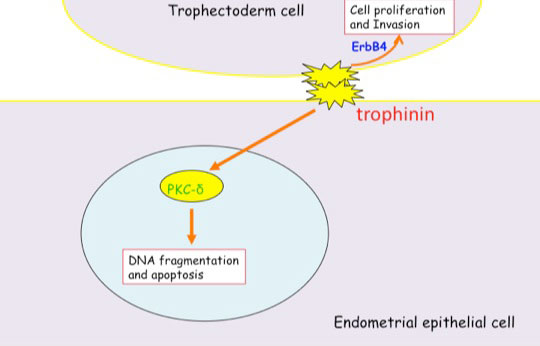
Distinct signal transduction of trophinin-mediated cell adhesion in trophectoderm cell and endometrial epithelial cells. In trophoblastic cells, ErbB4 (receptor tyrosine kinase) is arrested by bystin/trophinin complex. When trophinin-mediated cell adhesion takes palce, ErbB4 is released from bystin. This allows ErbB4 to be activated by phosphorylation. In endometrial epithelial cells, trophinin-mediated cell adhesion releases PKCd from trophinin. PKCd was then translocated to the nucleus, where it activates caspase 3 for apoptosis (Tamura et al., 2011).
References Cited
Hatakeyama S, Sugihara K, Shibata TK, Nakayama J, Akama TO, Tamura N, Wong SM, Bobkov AA, Takano Y, Ohyama C, Fukuda M, Fukuda MN (2011) Targeted drug delivery to tumor vasculature by a carbohydrate mimetic peptide. Proc Natl Acad Sci U S A 108: 19587-19592
Sugihara K, Kabir-Salmani M, Byrne J, Wolf DP, Lessey B, Iwashita M, Aoki D, Nakayama J, Fukuda MN (2008) Induction of trophinin in human endometrial surface epithelia by CGbeta and IL-1beta. FEBS Lett 582: 197-202
Sugihara K, Sugiyama D, Byrne J, Wolf DP, Lowitz KP, Kobayashi Y, Kabir-Salmani M, Nadano D, Aoki D, Nozawa S, Nakayama J, Mustelin T, Ruoslahti E, Yamaguchi N, Fukuda MN (2007) Trophoblast cell activation by trophinin ligation is implicated in human embryo implantation. Proc Natl Acad Sci U S A 104: 3799-3804
Tamura N, Sugihara K, Akama TO, Fukuda MN (2011) Trophinin-mediated cell adhesion induces apoptosis of human endometrial epithelial cells through PKC-delta. Cell Cycle 10 : 135-143