Scientists unearth a previously unknown vulnerability for cancer and a promising drug candidate that leverages the approach
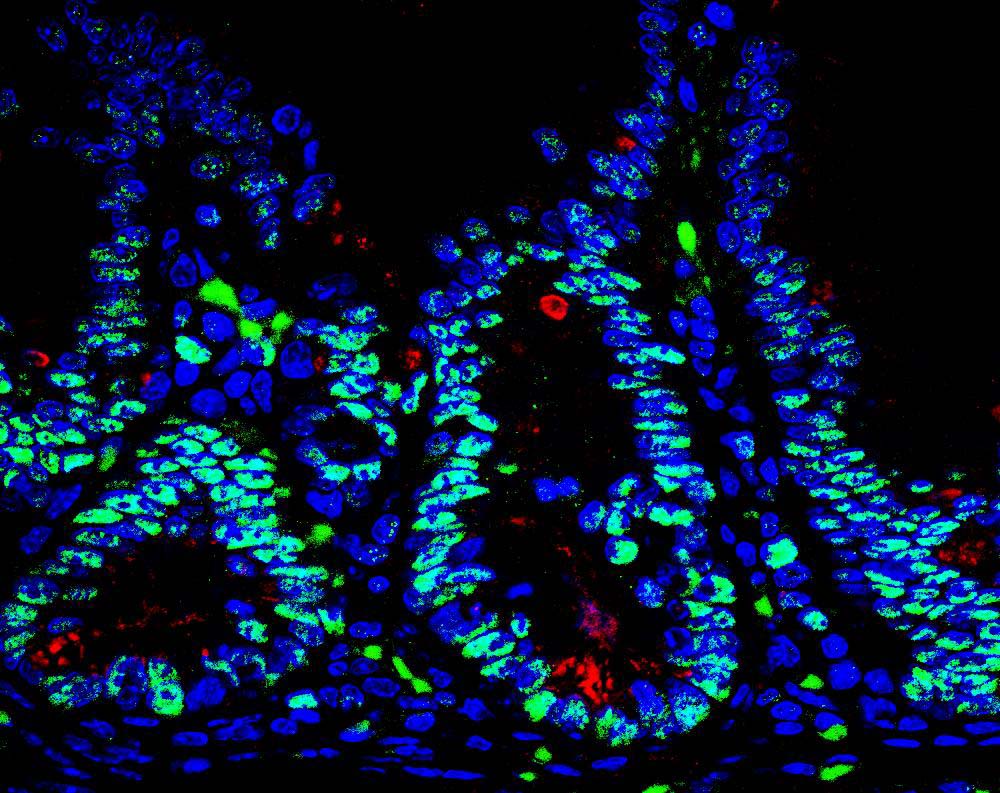
Scientists at Sanford Burnham Prebys have uncovered a drug candidate, called F5446, that exposes ancient viruses buried in “junk DNA” to selectively kill cancer cells. Published in the journal Cell, the proof-of-concept study reveals a previously unknown Achilles’ heel for cancer that could lead to treatments for deadly breast, brain, colon and lung cancers.
“We found within ‘junk DNA’ a mechanism to stimulate an immune response to cancer cells, while also causing tumor-specific DNA damage and cell death,” says Charles Spruck, PhD, assistant professor in the National Cancer Institute (NCI)-designated Cancer Center and senior author of the study. “This is a very new field of research, with only a handful of papers published, but this has the potential to be a game-changer in terms of how we treat cancer.”
Since the human genome was fully sequenced in 2003, scientists have learned that our DNA is filled with some very strange stuff—including mysterious, noncoding regions dubbed “junk DNA.” These regions are silenced for a reason—they contain the genomes of ancient viruses and other destabilizing elements. An emerging area of cancer research called “viral mimicry” aims to activate these noncoding regions and expose the ancient viruses to make it appear that a cancer cell is infected. The hypothesis is that the immune system will then be triggered to destroy the tumor.
A one-two punch to cancer
In the study, Spruck and his team set out to find the molecular machinery that silences “junk DNA” in cancer cells. Using sophisticated molecular biology techniques, they found that a protein called FBXO44 is key to this process. Blocking this protein caused the noncoding sections of DNA to unwind—but not for long.
“When we revealed noncoding regions, which aren’t meant to be expressed, this caused DNA breakage. This told the cell that something is deeply wrong, and it committed suicide,” explains Spruck. “At the same time, the DNA of the ancient virus was exposed, so the immune system was recruited to the area and caused more cell death. So, we really delivered a one-two punch to cancer.”
The scientists then showed that a drug that targets the FBXO44 pathway, called F5446, shrank tumors in mice with breast cancer. The drug also improved the survival of mice with breast cancer that were resistant to anti-PD-1 treatment, an immunotherapy that is highly effective but often stops working over time. Additional studies in cells grown in a lab dish showed that the drug stops the growth of other tumors, including brain, colon and lung cancers.
The scientists also conducted many experiments to show that this silencing mechanism only occurs in cancer cells, not regular cells. Analysis of patient tumor databases confirmed that FBXO44 is overproduced in many cancers and correlated with worse outcomes—further indicating that a drug that inhibits this protein would be beneficial.
Moving the research toward people
As a next step, the scientists are working with the Conrad Prebys Center for Chemical Genomics to design an FBXO44 pathway-inhibiting drug that is more potent and selective than F5446. This state-of-the-art drug discovery facility is located at Sanford Burnham Prebys.
“Now that we have a compound that works, medicinal chemists can make modifications to the drug so we have a greater chance of success when we test it in people,” says Jia Zack Shen, PhD, staff scientist at Sanford Burnham Prebys and co-first author of the study. “Our greatest hope is that this approach will be a safe and effective pan-cancer drug, which maybe one day could even replace toxic chemotherapy.”