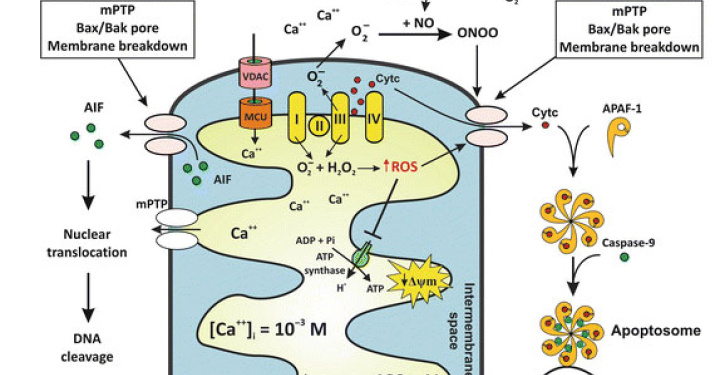
Related Disease: Neurodegenerative and Neuromuscular Diseases
Dr. Piña-Crespo earned a PhD in Pharmacology from University College London (UCL), England under the supervision of Profs. Alasdair Gibb & David Colquhoun FRS. He completed postdoctoral training as a Pew Fellow/Research Associate with Prof. Steve Heinemann in the Molecular Neurobiology Laboratory at The Salk Institute, La Jolla, California. Dr. Piña-Crespo has held faculty positions as Instructor and Assistant Professor at Universidad Centroccidental, Venezuela and as Lecturer in the Biology Department at the University of San Diego, California.
Education and Training
Postdoctoral training (Pew Fellow/Research Associate) The Salk Institute, California PhD in Pharmacology University College London (University of London), England Veterinarian (DVM) Universidad Centroccidental Lisandro Alvarado, Venezuela
Honors and Recognition
Pew Fellow in the Biomedical Sciences
Related Disease
Aging-Related Diseases, Alzheimer’s Disease, Brain Injury, Epilepsy, Molecular Biology, Nervous System Injury, Neurodegenerative and Neuromuscular Diseases, Neurological and Psychiatric Disorders, Parkinson’s Disease, Stroke, Traumatic Injury
Phenomena or Processes
Aging, Apoptosis and Cell Death, Calcium Signaling, Cell Biology, Cell Signaling, Cell Surface Receptors, Development of Neuronal Circuits, Disease Therapies, Neurobiology, Neurogenesis, Neuron-Glia Interactions in Myelin, Neurotransmitters, Synapse Function, Synaptic Transmission
Anatomical Systems and Sites
Brain, General Cell Biology, Nervous System
Research Models
Cultured Cell Lines, Human Cell Lines, Human Embryonic Stem Cells, Mouse, Mouse Cell Lines, Primary Cells, Primary Human Cells, Rat, Vertebrates, Xenopus
Techniques and Technologies
Biophysics, Biophysiology, Calcium Imaging, Cellular and Molecular Imaging, Electrophysiology, Fluorescence Microscopy, Ion Channels, Live Cell Imaging, Mouse Behavioral Analysis, Pharmacology, Transplantation
Working on basic neuroscience discovery research. I use cellular and animal models of neurodegeneration to identify basic disease-causing mechanisms and disease-relevant targets involved in abnormal neuron-glia signaling, synapse failure, neuronal network dysfunction and neuronal loss in age-related neurodegenerative diseases; including Alzheimer’s and Parkinson’s disease. Extensive hands-on experience working and managing projects that require a strong background in in-vitro, ex-vivo and in-vivo neuroscience, pharmacology and electrophysiology.
Anne Bang, PhD, focuses her research on bridging stem cell technology and drug discovery to enable the development of therapeutics. She was trained in developmental neurobiology and used both invertebrate and vertebrate models to study mechanisms that underlie spatial patterning of the nervous system in the developing embryo.
Bang’s experience with human pluripotent stem cells (hPSC) began in 2005 when she joined ViaCyte, Inc., as Director of Stem Cell Research, she led an interdisciplinary group developing hPSC-derived pancreatic cells to treat diabetes. Her efforts focused on optimizing differentiation processes and advancing Viacyte’s cell product into scaled manufacturing and studies enabling Investigational New Drug (IND) applications for the FDA. This work led to co-inventorship on multiple ViaCyte patents.
In 2010, Bang joined Sanford Burnham Prebys to lead hPSC-based disease modeling at the Conrad Prebys Center for Chemical Genomics (Prebys Center), a state-of-the-art drug discovery center. Her program leverages hPSC technology to generate disease-relevant models across different genetic backgrounds and differentiated lineages, enabling interrogation of cellular vulnerabilities and pathogenic phenotypes in human cells.
A central premise of Bang’s work is that these models are impactful when paired with quantitative, scalable phenotyping and systematic perturbation with small molecules, genetic modulation or other stressors to identify pathway-level control points and therapeutic hypotheses. Accordingly, her laboratory develops stem-cell-based assays that capture higher-order cellular and network functions while maintaining throughput compatible with discovery. Her team developed and ran an image-based, high-content screening platform in iPSC-derived neurons to identify chemical modulators of neurite outgrowth (Sherman and Bang, 2018), as well as a phenotypic screen in Alzheimer’s disease (AD) patient iPSC neurons that uncovered a druggable CYP46A1–cholesteryl ester–tau axis linking neuronal cholesterol metabolism to core AD pathologies (van der Kant et al., 2019).
To extend screening beyond morphology to systems-level function, she and her colleagues built multi-electrode array (MEA) workflows for iPSC-derived neuronal networks and have generated compound-response signatures across >270 perturbagens, including seizure-relevant phenotypes (in prep), alongside analytic methods to map network connectivity (Puppo et al., 2021). They further advanced MEA-based functional phenotyping by developing assays to model synaptic plasticity via long-term potentiation (LTP) (Pré et al., 2022) and to quantify oscillogenesis, emergent network dynamics and their pharmacological modulation, in scalable hPSC-derived cortical cultures (Pré et al., 2026).
Related Disease
Aging-Related Diseases, Alzheimer’s Disease, Amyotrophic Lateral Sclerosis (Lou Gehrig’s Disease), Congenital Disorders of Glycosylation, Glycosylation-Related Disorders, Multiple Sclerosis, Muscular Dystrophy, Neurodegenerative and Neuromuscular Diseases
Phenomena or Processes
Cell Biology, Development and Differentiation, Development of Neuronal Circuits, Embryonic/Pluripotent Stem Cells, Neurobiology, Neurodegeneration, Neurogenesis, Neuron-Glia Interactions in Myelin, Neurotransmitters, Synapse Formation and Maturation, Synapse Function, Synaptic Transmission
Research Models
Human Adult/Somatic Stem Cells, Human Cell Lines, Human Embryonic Stem Cells
Techniques and Technologies
Cellular and Molecular Imaging, Drug Discovery, Electrophysiology, Fluorescence Microscopy, High Content Imaging, In vivo Modeling, Microarrays, Molecular Genetics
Dr. Anne Bang is an experienced cell biologist and stem cell expert who leads efforts at the Prebys Center to develop patient cell specific and human induced pluripotent stem cell (hiPSC)-based disease models for drug screening and target identification. Her research program is primarily focused on neurological and neuromuscular disease, with the aim of designing human cell based models and assays that reflect higher order cellular functions and recapitulate disease phenotypes, yet have the throughput and reproducibility required for drug discovery. Towards this goal, her group has worked to develop a suite of foundational high throughput assays to monitor neuronal morphology, mitochondrial function and electrophysiology, using high content screening and multi-electrode array formats. They have conducted high-throughput drug screens on muscular dystrophy patient cells, hiPSC-derived cardiomyocytes and hiPSC-derived neurons, including from Alzheimer’s patient specific hiPSC. Bang is a principal investigator for the National Institute of Mental Health (NIMH) National Cooperative Reprogrammed Cell Research Groups consortium, and also receives research support from rare disease foundations and pharma sponsored collaborations.
Jackson, M., and Bang, A.G. 2013. Theranostics platform and methods of use, Patent Application No. PCT/US13/26448, Sanford Burnham Medical Research Institute, La Jolla, CA.
Ten scientists at Sanford Burnham Prebys Medical Discovery Institute were awarded eight grants yesterday from Curebound, a San Diego-based philanthropic organization
Guy Salvesen earned his PhD in biochemistry from Cambridge University in 1980. He conducted postdoctoral research at Strangeways Laboratory and MRC Laboratory of Molecular Biology in Cambridge, followed by further post-doctoral training at the University of Georgia. In 1991 he was appointed Assistant Professor at Duke University. Dr. Salvesen was recruited to Sanford-Burnham Medical Research Institute in 1996, where he is professor and director of the Apoptosis and Cell Death Research Program and dean of the Graduate School of Biomedical Sciences. He also holds an adjunct position as professor in the Department of Pathology at the University of California, San Diego.
Education
1981: PhD, Cambridge University, England, Biology 1977: B. Sc., London University, London, England, Microbiology
Other Appointments
Adjunct Professor, Department of Pathology, University of California, San Diego
Honors and Recognition
2014: Organizer, Keystone Meeting on Cell Death, February 2013: IUBMB Gold Medal Recipient, October 2010: Keynote Speaker, European Cell Death Organization Conference, 2010: Keynote Speaker, Gordon Research Conference on Cell Death 2009: Lifetime Achievement Award of the International Proteolysis Society 2008: Keynote Speaker, Queenstown Molecular Biology Conference 2008: Chair, Gordon Research Conference on Cell Death 2005: Helmut Holzer Memorial Prize 1999: International Proteolysis Society, Elected Secretary 1999: Keynote Speaker, Gordon Research Conference on Matrix Metalloproteinases 1988: American Association for the Study of Liver Diseases, State of the Art Lecture 1996: Chair, Gordon Research Conference on Proteolytic Enzymes and Their Inhibitors
Related Disease
Cancer, Inflammatory/Autoimmune Disease, Neurodegenerative and Neuromuscular Diseases, Pancreatic Cancer, Skin Cancer and Melanoma, Structural Biology
Phenomena or Processes
Apoptosis and Cell Death, Caspase Family, Cytokines, Inflammation, Protein Structure-Function Relationships, Proteolytic Pathways, Ubiquitin, Ubiquitin Protease System and Ubiquitin-like Proteins
Research Models
Bacteria, Human Cell Lines, Mouse, Mouse Cell Lines, Primary Human Cells
Techniques and Technologies
Biochemistry, Cellular and Molecular Imaging, Chemical Biology, Fluorescence Microscopy, Mass Spectrometry, Protein Engineering, Protein Structure Prediction, Protein-Protein Interactions, Proteomics
The human body contains cells with different life expectancies. Some (white blood cells or skin, for example) are programmed to rapidly die and be replaced. Others (such as nerve cells) are programmed to survive the lifetime of the individual and are seldom replaced. Dr. Salvesen’s research focuses on the central role enzyme pathways play in the life and death of cells. When death pathways slow down in cells that are normally programmed to die, cancer results. Conversely, when death pathways become overactive in cells that are programmed to survive, degenerative disease occurs. Dr. Salvesen’s laboratory focuses on understanding the fundamental molecular interactions that occur within these enzyme pathways. This knowledge is used to engineer synthetic compounds to stimulate cell destruction in cancer cells, or delay cell destruction in neurodegenerative diseases and stroke.
Guy Salvesen’s Research Report
Structure and Function of Proteases and Their Natural Inhibitors
Our research seeks to delineate the structure –> activity –> function algorithm as it applies to proteases and their inhibitors. Our laboratory has very broad interests in principles of proteolysis in humans, and we take multi-pronged approaches to research on proteases and their inhibitors.
Apoptosis
In one approach we apply basic biochemical knowledge to investigate newly emerging principles of proteolysis in human systems. This research is currently dissecting the proteolytic components of the intracellular pathway that lead to apoptotic cell death. Programmed cell death monitors the growth of new cells and the elimination of old ones. This program contains a number of proteolytic steps that are essential for efficient execution of the death pathway. Thus the proteases of the pathway – the caspases – are involved in the normal maintenance of correct cell number, and are therefore implicated in a number of pathologic and physiologic conditions. Using the techniques of protein chemistry, enzymology, crystallography, and recombinant DNA methodologies, we analyze the basic mechanism utilized by caspases to promote cell death pathways, and the mechanisms and specificity of the natural inhibitors that control them.
Cell Signaling
Modification of proteins by the small ubiquitin-like modifier SUMO is a dynamic and reversible process. The SUMO cycle begins when SUMO precursors are processed to remove short C-terminal extensions, thereby uncapping the C-terminal Gly-Gly motif that is essential for conjugation. SUMO ligases conjugate the protein, via its C-terminal carboxylate, to the side-chain lysine of target proteins to generate an isopeptide linkage. Eventually, SUMO is removed intact from its substrate SUMOylated proteins, and so the SUMOylation/deSUMOylation cycle regulates SUMOs function. A group of proteases known as SENPs are involved in both the activation of SUMO precursors (endopeptidase cleavage) and deconjugation of the targets (isopeptidase cleavage). Our laboratory is currently involved in projects to define the mechanisms that regulate SENP activity and access to their natural substrates.
Technology Development
The principle of proteolysis in vivo is to instigate irreversible changes to a set of protein substrates that alters their function and generates the required biological event. The sum total of the proteases and their target substrates operating in a physiologic pathway therefore defines the global event. Consequently, the identity of the substrate cleavages defines the proteases acting on them. We are developing proteomics-based methodologies, including selective protein labeling, multi-dimensional electrophoresis, and mass spectrometry techniques, to identify the products of proteolysis in vivo.
Dr. Tautz develops novel drugs targeting protein tyrosine phosphatases that are implicated in cancer, thrombosis, and Alzheimer’s disease.
Dr. Tautz earned his PhD in Organic Chemistry and Biochemistry from the University of Karlsruhe (Germany) with Dr. Janos Retey in 2002. He continued his research at the Burnham Institute with Dr. Tomas Mustelin, first as a postdoc and later as a staff scientist. In 2009 Dr. Tautz joined the faculty of the Sanford Burnham Prebys Medical Discovery Institute.
Education
2002: PhD, University of Karlsruhe, Germany, Chemistry, Biochemistry 1997: MS, University of Karlsruhe, Germany, Chemistry
Funding Awards and Collaborative Grants
American Heart Association Innovative Research Grant
Honors and Recognition
2014: Semifinalist, Stadtman Investigator Search, National Institutes of Health 2008/2006 : Society for Biomolecular Sciences Travel Awards 2006: American Chemical Society Travel Award 2006: William and Lillian Fishman Award for Exceptional Postdoctoral Research 2002: PhD in Chemistry magna cum laude, University of Karlsruhe, Germany
Related Disease
Alzheimer’s Disease, Cancer, Cardiovascular Diseases, Inflammatory/Autoimmune Disease, Leukemia/Lymphoma, Neurodegenerative and Neuromuscular Diseases
Many human diseases stem from abnormalities in the activities of protein kinases and protein phosphatases. While efficacious therapeutics targeting protein kinases have been successfully used in the clinic (e.g., Gleevec), effective strategies to target specific protein phosphatases are still elusive. Dr. Tautz’ laboratory works on novel, more effective approaches to target these important enzymes. He focuses on protein tyrosine phosphatases implicated in cancer, thrombosis, autoimmunity, and Alzheimer’s disease.
Lutz Tautz’s Research Report
1. Discovery of a Novel Drug Target in Arterial Thrombosis
Arterial thrombosis is the primary cause of most cases of myocardial infarction and stroke, the leading causes of death in the developed world. Platelets, highly specialized cells of the circulatory system, are key contributors to thrombotic events. Antiplatelet drugs, which prevent platelets from aggregating, have been very effective in reducing the mortality and morbidity of these conditions. However, approved antiplatelet therapies have adverse side effects, most notably the increased risk of bleeding. In collaboration with researchers at the University of Liege in Belgium (Drs. Souad Rahmouni and Cecile Oury), we recently identified DUSP3 (also known as VHR) as a major regulator in platelet signaling and thrombosis. Intriguingly, bleeding was not affected by DUSP3 deficiency in mice, suggesting that DUSP3 plays a key role in arterial thrombosis, but is dispensable for primary hemostasis. We develop a specific small-molecule inhibitor of DUSP3 that effectively inhibited platelet aggregation in human platelets, thereby phenocopying the effect of DUSP3 deficiency in murine platelets. We are now poised to optimize this compound for in vivo studies in order to provide proof-of-concept for a novel and potentially safer antiplatelet strategy based on DUSP3.
2. Discovery of a Novel Mechanism in T Cell Activation
PTPs are crucial for maintaining the homeostasis of the immune system, including the regulation of antigen receptor-mediated lymphocyte activation and cytokine-induced differentiation. The lymphoid tyrosine phosphatase (LYP, PTPN22) is a critical negative regulator of T cell antigen receptor signaling. A single-nucleotide polymorphism (SNP) in PTPN22 was shown to correlate with the incidence of various autoimmune diseases, including type 1 diabetes and rheumatoid arthritis. First, we helped to demonstrate that the disease-associated allele is a gain-of-function mutant, i.e. a better inhibitor of T cell receptor activation. Then, we developed a specific chemical probe of LYP which we utilized to identify the associated mechanism that leads to increased LYP activity. In contrast to what was known from work in mice, we showed that in human cells LYP needs to dissociate from CSK in order to inhibit T cell activation. We also showed that our LYP inhibitor acts by stabilizing a unique inactive conformation of LYP.
We have been working on chemical probes for cancer targets for several years (e.g., VHR, HePTP). Currently, we focus on the role of SHP2 in leukemia and breast cancer. PTPN11, the gene encoding SHP2, has been widely recognized as an oncogene. Germline mutations in PTPN11 were first observed in ~50% of cases of Noonan syndrome, an autosomal dominant developmental disorder with increased risk of malignancy. Numerous somatic gain-of-function mutations in PTPN11 have been identified in various leukemias. Hyperactivated SHP2 was also found in several types of solid tumors, including breast cancer. Previously reported SHP2 inhibitors lack efficacy in cancer cells and/or selectivity over related homologs. Novel SHP2 antagonists are needed for proof-of-principle studies that support a therapeutic approach based on SHP2. We have identified novel SHP2 lead compounds with significantly improved efficacy and selectivity. Currently, we optimize these compounds to make them suitable for in vivo studies.
Alzheimer’s disease (AD) is characterized by a progressive loss of cognitive function. The FDA has approved four drugs to treat the cognitive deficits in AD (donepezil, galantamine, rivastigmine, and memantine). However, none of these drugs halt disease progression. Our collaborator Dr. Paul Lombroso (Yale) identified the STriatal-Enriched Phosphatase (STEP) as a novel therapeutic target involved in the initial synaptic dysfunction that occurs prior to loss of neurons. His work suggests that inhibition of STEP could provide a disease-modifying strategy and early treatment option for AD. We recently received funding from the Alzheimer’s Association to develop a high-throughput assay to screen large chemical libraries for compounds that inhibit STEP function in neuronal cells. Once such compounds are identified, we will test their potential to reverse the biochemical and cognitive defects in AD animal models.
Douglas Sheffler studies the many facets of addiction, including addiction to the nicotine found in tobacco products. Smoking continues to be the leading cause of preventable deaths in the United States, and the second leading cause of preventable deaths worldwide after hypertension (of which smoking is a risk factor).
With Nicholas Cosford, PhD, Sheffler, in collaboration with colleagues at UC San Diego and Camino Pharma LLC, a San Diego-based biotechnology company Cosford co-founded, are conducting clinical trials to advance an investigational drug called SBP-9330.
SBP-9330 targets a neuronal signaling pathway that underlies addictive behaviors, including tobacco use. If ultimately approved for market, it would be a first-in-class oral therapeutic to help people quit smoking.
“Our research suggests that SBP-9330’s mechanism of action—how it works—may also be effective for other types of addiction, such as cocaine, opioid and methamphetamine. In the future, we hope to explore and broaden the drug’s therapeutic uses.”
Prior to coming to Sanford Burnham Prebys in 2012 as a research assistant professor, Sheffler served in the same capacity at Vanderbilt University. He earned his PhD in biochemistry from Case Western Reserve University and his Bachelor of Science degree from Saint Vincent College.
2013 NARSAD Young Investigator Award Brain and Behavior Research Foundation
Related Disease
Alzheimer’s Disease, Huntington’s Disease, Neurodegenerative and Neuromuscular Diseases, Neurological and Psychiatric Disorders, Schizophrenia
Dr. Sheffler joined the Sanford Burnham Prebys faculty in September 2012. Prior to this, he was a Research Assistant Professor at Vanderbilt University in the laboratory of P. Jeffrey Conn (2010-2012), where he also performed post-doctoral work (2006-2010). Dr. Sheffler has over 15 years’ experience in the study of G-protein coupled receptor (GPCR) signaling, assay development, high throughput screening for novel GPCR ligands, cell biology, neuroscience, and pharmacology. His interests are in the complex regulation of GPCRs: their signal transduction, ligand binding, receptor desensitization, and the processes of GPCR internalization and down-regulation. In addition, he has also has a specific interest in both the pharmacology of GPCRs, in general mechanisms of signal transduction, and in the pathogenesis of schizophrenia. During his graduate studies at Case Western Reserve University in the laboratory of Bryan L. Roth, MD, PhD, he discovered the regulation of 5-HT2A serotonin receptor signal transduction by p90 ribosomal S6 kinase (RSK2). During his post-doctoral studies and work as a Research Assistant Professor at Vanderbilt University, he focused on the discovery and characterization of orthosteric and allosteric modulators of GPCRs and led pharmacology efforts characterizing novel M1 muscarinic acetylcholine receptor agonists and antagonists, M1 positive allosteric modulators (PAMs), Glycine Transporter Type 1 (GlyT1) inhibitors, and novel Group II metabotropic glutamate receptor (mGlu) PAMs and NAMs. Dr. Sheffler’s research has resulted in more than 45 journal articles and is a listed inventor on five patent applications pertaining to small molecule therapeutics. Dr. Sheffler received a NARSAD Young Investigator Award in 2013 from the Brain and Behavior Research Foundation.
Douglas Sheffler’s Research Report
The metabotropic glutamate receptors (mGlus) are G protein-coupled receptors (GPCRs) that play numerous roles in modulating synaptic transmission and cell excitability. Recent preclinical and clinical studies provide strong evidence that agonists of the group II mGlus, comprised of the mGlu2 and mGlu3 subtypes, may provide a novel approach to treatment of schizophrenia and anxiety disorders. Based on this, there has been a major focus on understanding the roles of these receptors in regulating transmission in forebrain and limbic circuits. However, currently available orthosteric (glutamate site) agonists activate both mGlu2 and mGlu3 and do not provide insight into which subtype is most important for clinical efficacy. Alternatively, recent focus on compounds interacting with less highly conserved allosteric sites has led to advances in subtype selective compound development. Dr. Sheffler and others have discovered and characterized highly selective mGlu2 positive allosteric modulators (PAMs), that have no effect on mGlu3, and these compounds have allowed us to elucidate many of the physiological roles of mGlu2. These PAMs do not activate the receptor directly but act allosterically to potentiate glutamate responses. Dr. Sheffler and collaborators have also discovered group II mGlu negative allosteric modulators (NAMs) and have very recently discovered the first highly selective antagonist of mGlu3. The development of these pharmacological tools provides an opportunity to fully elucidate the roles of these mGlu subtypes. The group II mGlus play important roles in regulating transmission through the hippocampal formation. For example, activation of presynaptic group II mGlus reduces transmission at numerous hippocampal synapses including perforant path-dentate gyrus synapses and the mossy fiber synapse. In contrast, presynaptic group II mGlus are not involved in directly regulating transmission at the Schaffer collateral – CA1 (SC-CA1) synapse. However, we have previously reported extensive studies demonstrating group II mGlu involvement in a novel form of glial-neuronal communication in hippocampal area CA1. When coincidentally activated with β-adrenergic receptors (βARs) in astrocytes, group II mGlus induce a marked potentiation of cAMP responses elicited by activation of βARs. This synergistic increase in glial cAMP accumulation results in the release of adenosine, which activates presynaptic A1 adenosine receptors on neighboring SC terminals and induces a profound depression of transmission at the SC-CA1 synapse. This novel form of glial-neuronal signaling may provide a protective mechanism to reduce the risk of excitotoxicity when there is excessive excitatory drive to the hippocampus, such as during periods of intense or prolonged stress. This potential role has implications relevant for the therapeutic effects of group II mGlu agonists and is consistent with multiple studies suggesting group II mGlu agonists can reduce both acute and long term responses to stress. We have postulated that this effect is mediated by mGlu3 based on heavy expression of mGlu3 in hippocampal astrocytes. However, until now, selective reagents that differentiate between mGlu2 and mGlu3 have not been available to rigorously determine the specific group II mGlu subtype involved. The long term goal of Dr. Sheffler’s research is to establish the relative roles of individual group II mGlu subtypes in mediating glial-neuronal communication and modulating synaptic transmission in the hippocampus using pharmacological, biochemical, and electrophysiological approaches.
Dr. Yu Xin (Will) Wang received his PhD at the University of Ottawa where he identified cellular asymmetry and polarity mechanisms regulating muscle stem cell self-renewal and skeletal muscle regeneration. He then carried out postdoctoral training at Stanford University School of Medicine developing single cell multi-omic approaches to characterize the regenerative process and what goes awry with disease and aging.
“I’ve always had a passion for science and became fascinated with how the body repairs and heals itself when I was introduced to the potential of stem cells in regenerative medicine. I was struck by the ability of a small pool of muscle stem cells that can rebuild and restore the function of muscle. My lab at Sanford Burnham Prebys aims to better understanding the repair process and harness our body’s ability to heal in order to combat chronic diseases and even counteract aging.“
Education and Training
Postdoctoral Fellowship, Stanford University School of Medicine PhD in Cellular Molecular Medicine, University of Ottawa, Canada BS in Biomedical Sciences, University of Ottawa, Canada
Phenomena or Processes
Adult/Multipotent Stem Cells, Aging, Cell Signaling, Development and Differentiation, Epigenetics, Exercise, Extracellular Matrix, Neurogenesis, Organogenesis, Regenerative Biology, Transcriptional Regulation
Anatomical Systems and Sites
Immune System and Inflammation, Musculoskeletal System, Nervous System
Research Models
Clinical and Transitional Research, Computational Modeling, Human Adult/Somatic Stem Cells, Mouse
Techniques and Technologies
3D Image Analysis, Bioinformatics, Cellular and Molecular Imaging, Gene Knockout (Complete and Conditional), Genomics, High Content Imaging, High-Throughput/Robotic Screening, Live Cell Imaging, Machine Learning, Microscopy and Imaging, Proteomics, Transplantation
The Wang lab is interested in elucidating critical cell-cell interactions that mediate the function of tissue-specific stem cells during regeneration and disease, with a focus on how a coordinated immune response can promote regeneration and how autoimmunity impacts tissue function and hinder repair.
Specifically, the Wang lab aims to identify cellular and molecular crosstalk between muscle, nerve, and immune systems to develop targeted therapies that overcome autoimmune neuromuscular disorders and autoimmune aspects of “inflammaging.”
Yu Xin (Will) Wang’s Research Report
The lab’s research is translationally oriented and utilizes interdisciplinary molecular, genetic, computational (machine learning and neural networks), and bioengineering approaches to view biology and disease from new perspectives. We combine multi-omics sequencing and imaging methods to resolve how different cell types work together after injury to repair tissues and restore function. We use a data-driven approach to identify targetable disease mechanisms and, through collaborations with other researchers and clinicians, develop therapies that promote regeneration. Visit our lab website to learn more.
Sanford Burnham Prebys scientists say that understanding the potential pitfalls of using artificial intelligence and computational biology techniques in biomedical…
Evan Y. Snyder earned his MD and PhD (in neuroscience) from the University of Pennsylvania in 1980 as a member of NIH’s Medical Scientist Training Program (MSTP). He had also studied psychology and linguistics at the University of Oxford. After moving to Boston in 1980, he completed residencies in pediatrics and neurology as well as a clinical fellowship in Neonatal-Perinatal Medicine at Children’s Hospital-Boston, Harvard Medical School. He also served as Chief Resident in Medicine (1984-1985) and Chief Resident in Neurology (1987) at Children’s Hospital-Boston. In 1989, he became an attending physician in the Department of Pediatrics (Division of Newborn Medicine) and Department of Neurology at Children’s Hospital-Boston, Harvard Medical School. From 1985-1991, concurrent with his clinical activities, he conducted postdoctoral research as a fellow in the Department of Genetics, Harvard Medical School. In 1992, Dr. Snyder was appointed an instructor in neurology (neonatology) at Harvard Medical School and was promoted to assistant professor in 1996. He maintained lab spaces in both Children’s Hospital-Boston and at Harvard Institutes of Medicine/Beth-Israel Deaconess Medical Center. In 2003, Dr. Snyder was recruited to Sanford Burnham Prebys as Professor and Director of the Program in Stem Cell and Regenerative Biology. He then inaugurated the Stem Cell Research Center (serving as its founding director) and initiated the Southern California Stem Cell Consortium. Dr. Snyder is a Fellow of the American Academy of Pediatrics (FAAP). He also received training in Philosophy and Linguistics at Oxford University.
Related Disease
Alzheimer’s Disease, Amyotrophic Lateral Sclerosis (Lou Gehrig’s Disease), Arthritis, Brain Cancer, Brain Injury, Breast Cancer, Cancer, Childhood Diseases, Congenital Disorders of Glycosylation, HIV-Associated Dementia, Huntington’s Disease, Multiple Sclerosis, Muscular Dystrophy, Neurodegenerative and Neuromuscular Diseases, Neurological and Psychiatric Disorders, Parkinson’s Disease, Peripheral Vascular Disease, Skin Cancer and Melanoma, Spinal Cord Injury, Stroke, Traumatic Injury
We believe the study of stem cell biology will provide insights into many areas: developmental biology, homeostasis in the normal adult, and recovery from injury. Indeed, past and current research has already produced data in these areas that would have been difficult or impossible via any other vehicle. We have engaged in a multidisciplinary approach, simultaneously exploring the basic biology of stem cells, their role throughout the lifetime of an individual, as well as their therapeutic potential. Taken together, these bodies of knowledge will glean the greatest benefit for scientists and, most importantly, for patients. All of our research to date has been preformed in animal models with the ultimate goal of bringing them to clinical trials as soon as possible. Stem cells offer an intriguing mix of controversy, discovery, and hope. Politicians are charged with dealing with the controversial facets of stem cells, as we prefer to focus our energy on their potential for discovery and hope.
The Snyder Lab studies stem cell biology, with the goal of understanding normal development, tissue homeostasis, and recovery from injury and disease. A major focus is neural stem cells (NSCs), which can self-renew and differentiate into neurons, astrocytes, and oligodendrocytes. These properties make NSCs ideal for repair of damage due to injury or disease, but they also make them susceptible to transformation into malignant cancers.
Pier Lorenzo Puri earned his MD at the University of Rome “la Sapienza” in 1991. Dr. Puri completed his internship in Internal Medicine at the hospital “Policlinico Umberto I” (Rome) from 1992 to 1997, and defended an experimental thesis on the vascular effects of angiotensin II to graduate as Specialist in Internal medicine at the University of Rome “la Sapienza” in 1997. During this time he was frequently working at the Freien University of Berlin, as visiting scientist at the Deprtment of Biochemistry and Molecular Biology, to perform experiments of protein and DNA microinjection in cultured cells. Dr. Puri trained as a post-doctoral fellow at the University of California San Diego (UCSD), in the department of Cell Biology, under the supervision of Dr. Wang, from 1997 to 2001. He was appointed as Staff Scientist at the Salk Institute (La Jolla) in 2001, and became an Assistant Telethon Scientist at the Dulbecco Telethon Institute in Rome in 2002. He was upgraded to Associate Telethon Scientist at the Dulbecco Telethon Institute in Rome since 2007 and became Senior Telethon Scientist, Dulbecco Telethon Institute, in 2012, but declined this position. Dr. Puri joined Sanford Burnham Prebys as an Assistant Professor in 2004. He has been promoted to Associate Professor in 2010 and full Professor in 2015. From 2008 to 2016 Dr. Puri served as Adjunct Professor of Pediatrics at the University of California, San Diego. From 2008 to 2013 Dr Puri was an Associate Member of Sanford Children’s Health Research Center. Dr Puri has been Director of the laboratory of Epigenetics and Regeneration at Fondazione S. Lucia, Roma, Italy, but stepped down this position since 2019.
Education
University of California San Diego, Postdoctoral, Department of Biology University of Rome La Sapienza, PhD, Internal Medicine University of Rome La Sapienza, MD, Internal Medicine University of Rome La Sapienza, Undergraduate, Internal Medicine
Other Appointments
2020-2024: Member of the Science Advisory Board (SAB) European Commission-funded Consortium BIND (Brain Involvement In Dystrophinopathies) 2015-2019: Standing Member, NIH Study Section (SMEP) 2010-present: Member of Editorial Board of Skeletal Muscle
Phenomena or Processes
Adult/Multipotent Stem Cells, Aging, Cell Biology, Cell Cycle Progression, Cell Differentiation, Cell Signaling, Cellular Senescence, Development and Differentiation, Disease Therapies, DNA Damage Checkpoint Function, Epigenetics, Gene Regulation, Phosphorylation, Regenerative Biology, Signal Transduction, Transcriptional Regulation
Anatomical Systems and Sites
General Cell Biology, Musculoskeletal System
Research Models
Clinical and Transitional Research, Cultured Cell Lines, Human Adult/Somatic Stem Cells, Mouse Embryonic Stem Cells, Mouse Somatic Stem Cells, Primary Human Cells
Techniques and Technologies
Bioinformatics, Cellular and Molecular Imaging, Gene Expression, Genomics
Puri’s lab group investigates the molecular and epigenetic regulation of gene expression in skeletal muscle progenitors and other muscle-resident cell types (including fibro-adipogenic progenitors, cells from the inflammatory infiltrate, cellular components of neuro-muscular junctions) during physiological and pathological perturbations of skeletal muscle homeostasis.
We use molecular, biochemical and epigenetic tools to understand structural and functional principles of the 3D genome organization that regulates gene expression during muscle regeneration and diseases.
A topic of particular interest is the analysis of chromatin interactions that define the 3D genome organization and the identification of structural and functional interactions that regulate cell type-specific patterns of gene expression in response to cues released within the skeletal muscle regenerative environment in health and disease conditions, such as muscular dystrophies and other neuromuscular diseases.
The knowledge derived from our studies is instrumental to elucidate the pathogenesis of muscular disorders and discover pharmacological interventions that promote muscle regeneration to repair diseased muscles.
Current translational focus is devoted to:
the study of the therapeutic potential of HDAC inhibitors for treatment of Duchenne Muscular Dystrophy (DMD)
the identification of genome variants associated to DMD patient-specific patterns of expression of disease-modifier genes that can account for individual trends of disease progression beyond the common genetic deficiency of dystrophin
the effect of dystrophin deficiency and restoration by gene therapy on 3D genome and transcriptional output of DMD myofibers; the therapeutic potential of extracellular vesicles released by fibro-adipogenic progenitors of DMD skeletal muscles exposed to HDACi.
Puri Lab
Pier Lorenzo Puri’s Research Report
Puri’s lab group investigates the molecular and epigenetic regulation of gene expression in skeletal muscle progenitors and other muscle-resident cell types (including fibro-adipogenic progenitors, cells from the inflammatory infiltrate, cellular components of neuro-muscular junctions) during physiological and pathological perturbations of skeletal muscle homeostasis.
We use molecular, biochemical and epigenetic tools to understand structural and functional principles of the 3D genome organization that regulates gene expression during muscle regeneration and diseases
A topic of particular interest is the analysis of chromatin interactions that define the 3D genome organization and the identification of structural and functional interactions that regulate cell type-specific patterns of gene expression in response to cues released within the skeletal muscle regenerative environment in health and disease conditions, such as muscular dystrophies and other neuromuscular diseases.
The knowledge derived from our studies is instrumental to elucidate the pathogenesis of muscular disorders and discover pharmacological interventions that promote muscle regeneration to repair diseased muscles
Current translational focus is devoted to:
the study of the therapeutic potential of HDAC inhibitors for treatment of Duchenne Muscular Dystrophy (DMD)
the identification of genome variants associated to DMD patient-specific patterns of expression of disease-modifier genes that can account for individual trends of disease progression beyond the common genetic deficiency of dystrophin
the effect of dystrophin deficiency and restoration by gene therapy on 3D genome and transcriptional output of DMD myofibers; the therapeutic potential of extracellular vesicles released by fibro-adipogenic progenitors of DMD skeletal muscles exposed to HDACi.
1. Epigenetic regulation of skeletal myogenesis by histone acetyltransferases and deacetylases
Our earlier identification and characterization of acetyltransferases p300/CBP and PCAF, as transcriptional co-activators, and the histone deacetylases HDACs, as transcriptional co-repressors, of the myogenic determination factor MyoD1-3, respectively, inspired the experimental rationale toward exploiting pharmacological inhibition of HDAC to promote skeletal myogenesis.
2. HDAC inhibitors as pharmacological intervention in DMD and other muscular dystrophies
Puri lab discovered that dystrophin-activated nNOS signalling controls HDAC2 activity, thereby revealing a previously unrecognized link between constitutive activation of HDAC2 and alteration of the epigenetic landscape of dystrophin-deficient muscles6,7. This discovery established the rationale for using HDAC inhibitors to counter the progression of Duchenne muscular dystrophy (DMD), by correcting aberrant HDAC activity in dystrophin-deficient muscles8-11.
3. Control of chromatin structure in muscle cells by regeneration-induced signaling pathways
Upon the discovery and characterization of intracellular signaling pathways (i.e. p38, ERK and AKT cascades) that regulate muscle gene expression in myoblasts, in earlier studies during Puri’s postdoctoral training, Puri lab has revealed the mechanism by which muscle environmental cues are converted into epigenetic changes that regulate gene expression in healthy and diseased muscles, via extracellular signal-activated kinase targeting of chromatin-modifying enzymes. These studies provided the first evidence that regeneration activated p38 and AKT signaling cooperatively direct assembly and activation of histone acetyltransferases and chromatin remodeling SWI/SNF complex at myogenic loci in muscle progenitors12,13,15. Moreover, we discovered that regeneration-activated p38 targets Polycomb Repressory Complex (PCR2) at Pax7 locus to promote formation of repressive chromatin during satellite cells a ctivation14.
4. Epigenetic basis for activation of the myogenic program in ESCs and other pluripotent cell types
Puri lab studied the epigenetic determinants of human embryonic stem cells (hESCs) and induced pluripotent stem cells (hiPSCs) commitment to skeletal myogenesis, by investigating the hESC resistance to direct conversion into skeletal muscle upon ectopic expression of MyoD, which can otherwise reprogram somatic cells into the skeletal muscle lineage. These studies showed that hESC and hiPSC resistance to myogenic conversion is caused by the lack of expression of one structural component of the SWI/SNF chromatin remodelling complex – BAF60C – which is specifically induced in embryoid bodies13. Based on these studies, we have recently established a protocol of hESC-derived 3D contractile myospheres that offers the unprecedented opportunity to dissect and analyze the epigenetic dynamics that underlie the formation of skeletal muscles and to identify changes in the epigenome induced by contractile activity in healthy vs dystrophin-deficient myofibers16,20. We have also determined the identity of the general transcription factors implicated in the activation of skeletal myogenesis17, and we have discovered that replicative senescence is associated with acquisition of resistance to MYOD-mediated activation of muscle gene expression, caused by the constitutive activation of DNA damage repair (DDR) response that impairs cell cycle progression and MYOD activity18. Finally, our recent work has elucidated the mechanism by which MYOD regulates high-order chromatin interactions to define the tri-dimensional (3D) nuclear architecture for the activation of skeletal myogenesis during human somatic cell reprogramming into skeletal muscles19.
5. Identification, functional, phenotypic and molecular characterization of muscle-interstitial cells – (the fibroadipogenic progenitors – FAPs) in healthy and diseased muscles.
Our work has elucidated the molecular determinants of the interplay between adult muscle stem cells and cellular components of their functional niche (i.e. FAPs), by identifying regulatory networks implicated in compensatory or pathogenic regeneration, and suggesting “disease stage-specific” responses to pharmacological treatment of neuromuscular disorders, such as DMD. Indeed, we have shown that HDACi promote compensatory regeneration and prevent fibro-adipogenic degeneration in mdx mice at early stages of diseases, by targeting a population of muscle interstitial cells – FAPs8 – and have identified a HDAC-regulated network that controls expression of myomiRs and alternative incorporation of BAF60 variants into SWI/SNF complexes to direct the pro-myogenic or fibro-adipogenic FAP activity21. Furthermore, we have recently identified specific subpopulations of FAPs (subFAPs) in physiological conditions and disease22 and we have discovered that specific subFAPs expand and adopt pathogenic phenotypes upon muscle denervation23 or in muscles of patients affected by type 2 diabetes24.
Elena Pasquale earned her PhD in biology from the University of Parma, Italy. She did postdoctoral work at Cornell University, after which she was appointed Research Assistant Professor at University of Parma. Following a second postdoctoral training period at the University of California in San Diego, Dr. Pasquale was appointed Assistant Research Biologist at that institution. Dr. Pasquale was recruited to Sanford Burnham Prebys in 1990.
Related Disease
Cancer, Neurodegenerative and Neuromuscular Diseases, Skin Cancer and Melanoma
Cancer, Neurodegenerative and Neuromuscular Diseases
Receptor tyrosine kinases of the Eph family and their ligands, the ephrins, represent an important cell communication system that controls a vast array physiological and disease processes. For example, Eph receptors and ephrins take part in the formation of blood vessels, including the blood vessels that feed tumors, and regulate the malignant properties of cancer cells and their interplay with the tumor microenvironment. They also regulate the formation, plasticity and regeneration of neuronal circuits as well as neurodegenerative processes such as those occurring in amyotrophic lateral sclerosis (ALS) and Alzheimer’s disease. The signal transduction mechanisms of Eph receptors are intriguing, and complex, because these receptors engage in multiple modes of signaling. Binding to ephrin ligands on the surface of neighboring cells induces canonical signaling involving receptor clustering, autophosphorylation on tyrosine residues, and kinase activity-dependent downstream signaling. Binding to the Eph receptors can also cause the ephrins, which have a cytoplasmic domain or a GPI-anchor, to transmit signals. This leads to bidirectional signals emanating from Eph receptor-ephrin complexes positioned at sites of cell-cell contact. In addition, at least some Eph receptors can also signal through non-canonical mechanisms that are independent of ligand binding and kinase activity, for example through interplay with other receptor tyrosine kinase families and with serine/threonine kinases.
Our research investigates Eph receptor signaling activities in order to understand their role in normal physiology and in pathological conditions such as cancer and neurodegenerative disorders. This knowledge is useful for the development of disease treatments based on modulating Eph receptor/ephrin activities. Ongoing efforts in our laboratory also focus on the development of agents targeting Eph receptors for research and translational applications.
Elena Pasquale’s Research Report
We discovered several Eph receptors and ephrins, and research in our laboratory is dedicated to the characterization of Eph receptor signal transduction mechanisms and biological functions using biochemical, mass spectrometry, molecular biology and cell biology approaches in conjunction with animal models. We have identified tyrosine and serine/threonine phosphorylation sites of Eph receptors and ephrins using mass spectrometry and investigated the signaling role of these phosphorylation sites. For example, our past work showed that two conserved tyrosine phosphorylation sites in the juxtamembrane segment of the Eph receptors not only mediate association with binding partners but also regulate receptor kinase activity. We also found that the SRC and ABL non-receptor tyrosine kinases and the SHEP1 scaffolding protein are binding partners of the Eph receptors, and we identified signaling connections between Eph receptors and integrins. We also found that EphA4 is highly expressed in the adult brain, where it regulates synaptic connections. More recent work in our laboratory focuses on elucidating signaling pathways that mediate the activities of Eph receptors in cancer cells.
Tumor Suppression and Tumor Promotion by Eph Receptors
Many Eph receptors are highly expressed in tumors, but their role in cancer is incompletely understood and likely depends on the cellular context. Certain Eph receptors and ephrins promote tumor angiogenesis. We showed that the EphA2 receptor is upregulated in the tumor vasculature together with the ephrin-A1 ligand, which suggested a role in tumor angiogenesis that is now well established. We also found that the EphB4 receptor expressed on the surface of breast cancer cells can promote tumor xenograft growth by enhancing blood vessel formation through interactions with its preferred ligand, ephrin-B2, present in tumor endothelial cells. Additional intriguing roles for the Eph receptors in cancer progression have also emerged. We found that canonical signaling by the EphB4 receptor is low in breast cancer cells and that ephrin-induced stimulation of EphB4 kinase activity inhibits breast cancer cell malignancy in culture and tumor growth in vivo (Figure 1A) through inhibition of the CRK proto-oncogene. More recently, we elucidated an additional mechanism of tumor suppression mediated by canonical ephrin-induced EphA2 signaling (Figure 1A), which leads to inhibition of the AKT-mTORC1 oncogenic pathway through interplay of EphA2 with a phosphatase that dephosphorylates the AKT serine/threonine kinase.
Figure 1. Dual activities of Eph Receptors in Cancer Cells. (A) Eph receptor-ephrin binding at cell-cell contact sites results in the dimerization/clustering of Eph receptor-ephrin complexes, and initiation of canonical signals through the receptor cytoplasmic domain. Signals through the ephrins can also be generated. Tyrosine phosphorylation sites (yellow circles) promote Eph kinase activity and also provide binding sites for signaling proteins containing SH2 domains. Other effectors also mediate Eph signals, including PDZ domain-containing proteins. The Eph receptor domains are indicated on the left; LBD, ligand-binding domain. (B) Eph receptors can potentiate the oncogenic effects of other receptors. These activities are independent of ephrin binding and/or kinase activity and their mechanism is not well understood but in some cases depends on Eph receptor phosphorylation on serine/threonine residues (red circle).
There is also evidence that some Eph receptors can increase cancer cell malignancy through non-canonical ephrin-independent and/or kinase-independent signaling activities (Figure 1B), which is the subject of ongoing work. These tumor promoting activities include inducing invasiveness and metastasis, epithelial-to-mesenchymal transition, stem cell-like features and drug resistance.
Eph Receptor Mutations in Cancer
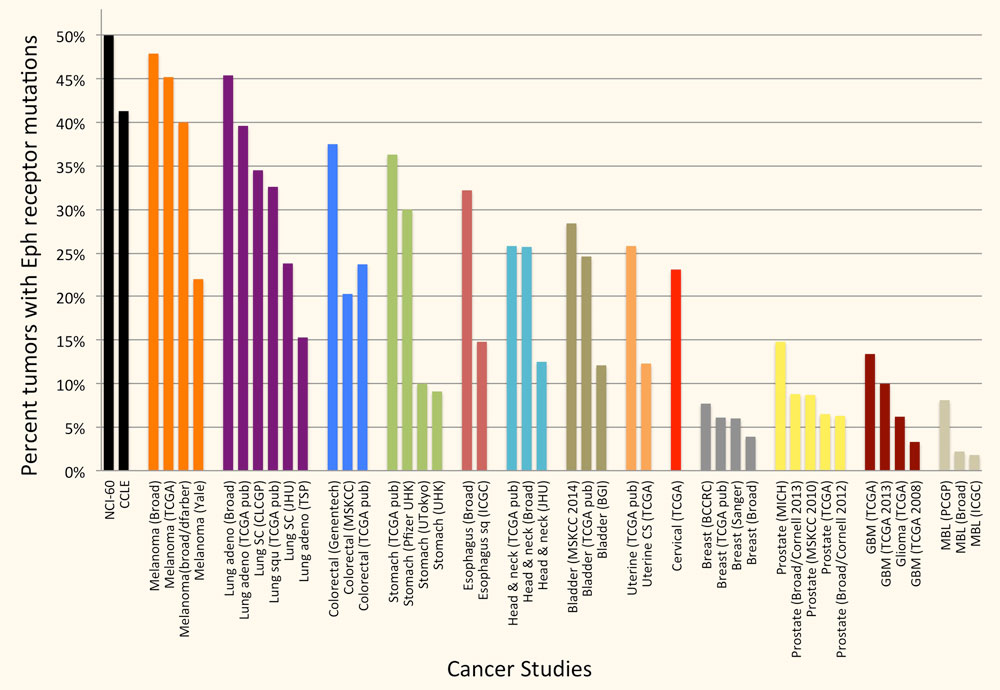
The Eph receptors are frequently mutated in many types of cancer. In particular, genome-wide sequencing studies have detected somatic mutations in one or more Eph receptors in 25%-45% of melanomas, 15%-45% of lung cancers, 25-40% of colorectal cancers and 12%-25% of head and neck and uterine cancers (Figure 2), but very limited information is available on the effects of the mutations. Studies by ours and other groups have shown that a number of EphA2 and EphA3 mutations inactivate Eph receptor canonical signaling by disrupting ephrin binding or kinase activity, consistent with a role of canonical signaling in tumor suppression. Ongoing work in our laboratory focuses on characterizing the functional effects of Eph receptor mutations in cancers such as melanoma, and investigating whether the mutations shift the balance of the Eph receptor signaling activities from tumor suppression to tumor promotion. We are also interested in the interplay of Eph receptor mutations with mutations affecting well-established oncogenes and tumor suppressor genes. Understanding the effects of Eph receptor mutations in cancer cells will help shed light on the role of the Eph receptor/ephrin system in cancer cell transformation, malignant progression and drug resistance.
Figure 2. A large percentage of tumor specimens and cell lines harbor one or more Eph receptor mutations. Groups of bars of the same color represent studies of the same cancer type. The cancers with most Eph receptor mutations are shown; other tumor types have fewer or no Eph receptor mutations. The graph is based on data from cBioPortal for Cancer Genomics (www.cbioportal.org).
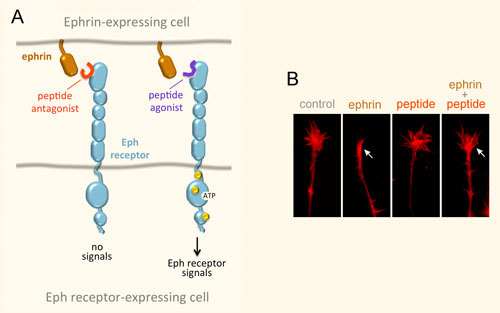
Peptides Targeting Eph Receptors
We have identified a number of peptides that bind to Eph receptors and inhibit ephrin binding by using phage display approaches. Collaborating groups have elucidated the structural features of several of these peptides in complex with the ligand-binding domain of Eph receptors, demonstrating that the peptides bind to the ephrin-binding pocket in the ligand-binding domain (Figure 3A). Most of the peptides are antagonists, but the peptides targeting EphA2 are agonists that activate receptor signaling and endocytosis similarly to the natural ephrin ligands. Interestingly, some of the identified peptides are highly specific and bind to only one Eph receptor family member. This is unlike the natural ephrin ligands, each of which promiscuously binds to multiple Eph receptors. Thus, Eph receptor-targeting peptides represent valuable pharmacological tools to study the functional importance of specific Eph receptors in tumors and the nervous system. Furthermore, they could be used as leads to develop therapies against cancer and neurological disorders, and to promote neural repair after nervous system injury (Figure 3B). Finally, our peptides have been used by other groups to deliver conjugated imaging agents, drugs and nanoparticles to Eph receptor-positive tumors. Current work focuses on identifying novel Eph receptor-targeting agents (such as peptides and small molecules) as well as improving the existing ones in collaboration with medicinal chemists and structural biologists, and evaluating them in cell culture and in vivo animal models.
Figure 3. Peptides can target the ephrin-binding pocket of Eph receptors with high affinity and specificity, affecting receptor function. (A) Peptides targeting the Eph receptors can function as antagonists that inhibit ephrin binding and receptor signaling, or in some cases as agonists that mimic the ephrins by activating Eph receptor signaling. Yellow circles indicate tyrosine phosphorylation sites in the activated Eph receptor. (B) An EphA4 peptide antagonist blocks ephrin-induced growth cone collapse in EphA4-expressing axons, suggesting its usefulness for promoting neural repair. The arrow in the second panel marks a growth cone collapsed due to ephrin treatment; the arrow in the fourth panel marks a growth cone that did not collapse following ephrin treatment in the presence of a peptide antagonist.
Nicholas Cosford, PhD has served on the Sanford Burnham Prebys Board of Trustees since 2023. He is the first faculty member to do so.
Cosford joined the Sanford Burnham Prebys faculty in 2008 as an associate professor. In 2013, he became a full professor. His lab investigates the interactions of small molecule compounds with therapeutically important proteins and cellular signaling pathways. With a specific focus on the discovery and optimization of compounds that might treat cancer, central nervous system diseases and infectious diseases.
Prior to joining Sanford Burnham Prebys in 2005, Cosford worked in both the biotechnology and pharmaceutical industries. At Sibia Neurosciences and at Merck Research Laboratories, he directed multidisciplinary research teams focused on small-molecule hit-to-lead optimization and was responsible for moving several lead compounds through to the clinical phase, including a nicotinic agonist (Altinicline) from research to Phase II clinical trials for treating Parkinson’s disease.
He is an author of more than 90 peer-reviewed, published scientific papers, and has been issued more than 40 issued patents, with an additional 40 patent applications pending.
Cosford has a Bachelor of Science degree in chemistry from the University of Bath in England and Doctor of Philosophy degree in organic chemistry from Emory University in Atlanta, GA.
Related Disease
Alzheimer’s Disease, Amyotrophic Lateral Sclerosis (Lou Gehrig’s Disease), Bone Mineralization Disorders, Breast Cancer, Cancer, Neurodegenerative and Neuromuscular Diseases, Neurological and Psychiatric Disorders, Ovarian Cancer, Pancreatic Cancer, Prostate Cancer
We are interested in investigating the interactions of small molecule compounds with therapeutically important proteins and cellular signaling pathways. One aspect of our research emphasizes the use of medicinal chemistry and chemical biology approaches to probe intracellular pathways that regulate cell survival and cell growth. Another area of active research is the development of synthetic chemistry methodology using microfluidic technology for the rapid synthesis of biologically active small molecules. Therapeutically, we are primarily focused on the discovery and optimization of compounds that have the potential to treat cancer, CNS diseases and infectious diseases.