Manuel Perucho earned his PhD in biological sciences at the University of Madrid, Spain in 1976. He did postdoctoral work at the Max-Planck-Institut für Molekulare Genetik, Berlin and at Cold Spring Harbor Laboratory, where he was subsequently appointed to staff in 1981. Following appointments at SUNY Stony Brook as Assistant and Associate Professor in 1982 and 1987, respectively, Dr. Perucho joined the California Institute for Biological Research in La Jolla, serving as Research Program Director from 1993 to 1995. Dr. Perucho was recruited to Sanford Burnham Prebys in 1995.
Other Appointments
Adjunct Professor, Pathology Department, University of California, San Diego
Related Disease
Colorectal Cancer, Endometrial Cancer, Gastric Cancer, Ovarian Cancer
Dr. Perucho studies tumors from the intestinal tract that sometimes develop when the cellular machinery preserving the integrity of the genome – like computer spell-check programs that detect errors and correct them – is not working properly. When these corrector genes (mutators) are inactivated, the mutations that occur in all normal cells accumulate in large numbers because they are not repaired. This sparks genomic instability and cancer eventually develops when mutations occur in some cancer genes, such as oncogenes and tumor suppressor genes. However, some mutator genes are not inactivated by mutations, but by epigenetic silencing. This results from the disintegration of the epigenetic code, an unexplored process that is strongly associated with aging. This is important because many hereditary colon tumors originate by mutations in mutator genes that are transmitted from generation to generation. Molecular diagnosis of the deficient mutator genes determines which members of these families will be affected in the future. Identification of tumors with this kind of genomic instability is also useful for detecting familial cancer patients and predicting survival.
Manuel Perucho’s Research Report
Genomic Instability in Cancer Pathways
Our research efforts focus on the analysis of the genomic instability underlying two alternative pathways for oncogenesis (see figure below). Most neoplasms lose the chromosomal balance of the diploid normal cell following a pathway for cancer that involves the mutational inactivation of critical tumor suppressor genes. A minority of cancers manifest another type of genomic instability – the accumulation of hundreds of thousands of mutations, including insertions and deletions of a few base pairs in simple repeated sequences or microsatellites.
We study the aneuploidy of the tumor cell of the suppressor pathway for cancer by unbiased Arbitrarily Primed PCR DNA fingerprinting. Gains and losses of sequences from defined chromosomal regions can be simultaneously identified in multiple tumors generating a molecular karyotype or “amplotype.” Amplotyping offers useful applications for cancer diagnosis and prognosis and maps chromosomal regions harboring cancer genes with positive and negative roles in cell growth or survival. These cancer genes are under positive and negative selection pressure during tumorigenesis and are detected by the frequent gains and losses of specific chromosomal regions, respectively.
Understanding Colon Cancer
In colon cancer, the initial event in this carcinogenic pathway is the inactivation of the APC tumor suppressor. In hereditary cases of the familial polyposis (FAP) cancer syndrome, one mutated allele is transmitted in the germline, while in sporadic cases both alleles are inactivated by somatic mutations. Usually one allele is inactivated by a nonsense or frameshift mutation and the other allele is inactivated by the deletion of the chromosomal region (loss of heterozygosity), typical of the aneuploid cancer cell.
The first event in the Microsatellite Mutator Phenotype (MMP) pathway for colon cancer is the inactivation of a gene involved in genome stability, such as the hMLH1 DNA mismatch repair gene. In sporadic cases, the inactivation of the mutator gene usually occurs by somatic mutations or by epigenetic silencing. In many familial cases, including a majority of the Hereditary Non-Polyposis Colorectal Cancer (HNPCC) syndrome, one allele is inactivated by a germline mutation and the other by any of the other mechanisms (mutation, LOH, epigenetic silencing, etc.).
The MMP pathway for gastrointestinal cancer presents two distinctive features that seem paradoxical at first sight. First, despite accumulating hundreds of thousands of clonal somatic mutations in simple repeated sequences, these tumors exhibit a low mutation incidence in APC, K-ras and p53, prototypical cancer genes in colorectal carcinogenesis. Second, these tumors harbor ubiquitous biallelic mutations in non-functional poly (A)n sequences, such as the poly A tails of the Alu repeats. However, they also accumulate many monoallelic (i.e., heterozygous) mutations in functional sequences, such as the coding regions of mutator (hMSH3, hMSH6), suppressor (TGFbRII, p53) and apoptotic (Bax) genes.
The first paradox may be explained by the existence within some genes of simple repeats that are preferred targets for the MMP. Thus, in the presence of the mutator phenotype, mutations in these genes (i.e., Bax) occur sooner than in other genes of the same oncogenic signaling pathways that do not have these repeats (i.e., p53). The second paradox can also be explained by another peculiar feature of these MMP tumors. Due to their exacerbated mutator phenotype, the disruption of the homeostatic controls for cell growth and survival may also occur by the accumulation of heterozygous mutations in multiple genes whose products play redundant but synergistic roles at different points of the cell proliferation and apoptotic networks. The occurrence of multiple heterozygous mutations presumably reduces the threshold amounts of the corresponding gene products. This accumulative haploinsufficiency model is not restricted to cell proliferation and apoptotic pathways, but also applies to other networks involved in the control of genome integrity.
Michiko N. Fukuda earned her PhD in biochemistry at the University of Tokyo in 1980. She did postdoctoral work at Fred Hutchinson Cancer Research Center in Seattle prior to her recruitment to Sanford-Burnham Medical Research Institute in 1982.
Education
1980: PhD, University of Tokyo, Biochemistry 1970: MS, University of Tokyo, Biochemistry 1968: BS, Tokyo University of Education, Botany
Related Disease
Breast Cancer, Cancer, Congenital Disorders of Glycosylation, Endometriosis, Glycosylation-Related Disorders, Inherited Disorders, Ovarian Cancer, Prostate Cancer, Testicular Cancer
Michiko Fukuda’s Research Report
Identification of Peptide that Delivers Drugs to Tumors
Chemotherapy effectiveness is often limited by drug toxicity in healthy tissues, although methods that spare normal cells by delivering drugs specifically to tumors may help to overcome this constraint. We have identified a promising tumor-targeted drug delivery vehicle known as the IF7 peptide. Using in vitro assays, we found that the IF7 peptide bound to the protein annexin 1 (Anxa1), which is known from previous studies by others to be enriched on the surface of tumor vasculature in several tumor types. When we injected a fluorescently labeled IF7 peptide into mice with tumors, fluorescent signals appeared in the tumors within one minute of injection. By contrast the tumors showed no fluorescence when mice were pre-injected with anti-Anxa1 antibodies that inhibits IF7-Anxa1 binding, suggesting that the peptide targets tumor by homing to Anxa1.
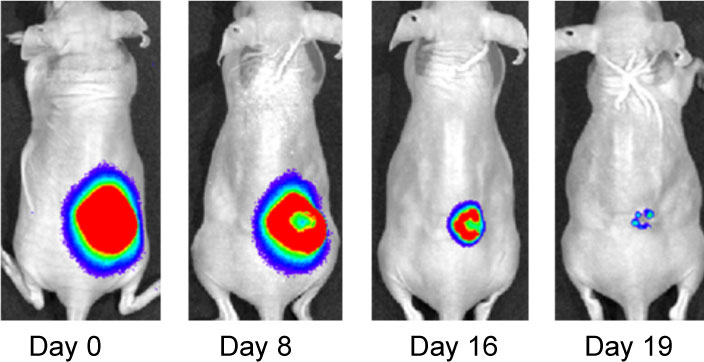
Effect of IF7-conjugated anti-cancer drug SN-38 on colon cancer mouse model. Activa cancer cells in a large tumor produce chemiluminescence which is captured by imaging. (Hatakeyama et al, 2011).
When IF7 peptide was conjugated with potent anti-cancer drug SN-38 and IF7-SN38 conjugate was injected intravenously to mice with tumors, IF7 could deliver SN-38 to tumors. We found that daily injections of an IF7-SN38 conjugate reduced a large tumor in the mouse without apparent side effects, whereas non-homing peptide-SN38 conjugate or with SN-38 alone did not reduce the tumors (Hatakeyama et al, 2011). The findings suggest that IF7 peptide may represent a clinically relevant vehicle for anti-cancer drugs.
Role of Trophinin in Human Embryo Implantation and Cancer
Invasion of the trophoblast into the endometrium, an essential element of embryo implantation, resembles invasion of malignant tumors. At the initial phase of implantation, the trophoblast and the uterine epithelium establish their first contact via their respective apical cell membranes. We have identified new molecules, trophinin, tastin, and bystin that mediate cell adhesion between trophoblastic cells and endometrial epithelial cells at the respective apical cell membranes. Trophinin is an intrinsic membrane protein, and tastin and bystin are cytoplasmic proteins. All of these molecules are strongly expressed in cells involved in embryo implantation in humans. However, trophinin is not expressed in human endometrial epithelia throughout the hormonal cycle, except only those cells located close to the implanting blastocyst. Trophinin expression by endometrial epithelia is induced by human chorionic gonadotrophin (hCG) derived from the implanting embryo (Sugihara et al, 2008). While embryos invade maternal cells (Sugihara et al, 2007), maternal tissue accepts embryos. We asked what happens in the maternal epithelia when trophinin-mediated adhesion takes place, and found that trophinin-mediated cell adhesion triggers an apoptotic signal in maternal epithelial cells (Tamura et al, 2011).
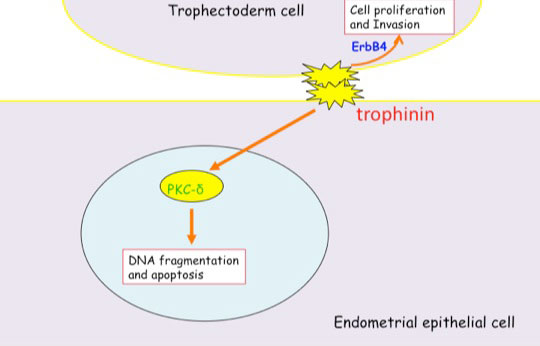
Distinct signal transduction of trophinin-mediated cell adhesion in trophectoderm cell and endometrial epithelial cells. In trophoblastic cells, ErbB4 (receptor tyrosine kinase) is arrested by bystin/trophinin complex. When trophinin-mediated cell adhesion takes palce, ErbB4 is released from bystin. This allows ErbB4 to be activated by phosphorylation. In endometrial epithelial cells, trophinin-mediated cell adhesion releases PKCd from trophinin. PKCd was then translocated to the nucleus, where it activates caspase 3 for apoptosis (Tamura et al., 2011).
References Cited
Hatakeyama S, Sugihara K, Shibata TK, Nakayama J, Akama TO, Tamura N, Wong SM, Bobkov AA, Takano Y, Ohyama C, Fukuda M, Fukuda MN (2011) Targeted drug delivery to tumor vasculature by a carbohydrate mimetic peptide. Proc Natl Acad Sci U S A 108: 19587-19592
Sugihara K, Kabir-Salmani M, Byrne J, Wolf DP, Lessey B, Iwashita M, Aoki D, Nakayama J, Fukuda MN (2008) Induction of trophinin in human endometrial surface epithelia by CGbeta and IL-1beta. FEBS Lett 582: 197-202
Sugihara K, Sugiyama D, Byrne J, Wolf DP, Lowitz KP, Kobayashi Y, Kabir-Salmani M, Nadano D, Aoki D, Nozawa S, Nakayama J, Mustelin T, Ruoslahti E, Yamaguchi N, Fukuda MN (2007) Trophoblast cell activation by trophinin ligation is implicated in human embryo implantation. Proc Natl Acad Sci U S A 104: 3799-3804
Tamura N, Sugihara K, Akama TO, Fukuda MN (2011) Trophinin-mediated cell adhesion induces apoptosis of human endometrial epithelial cells through PKC-delta. Cell Cycle 10 : 135-143
Kristiina Vuori earned her MD and PhD at University of Oulu, Finland. After completion of internship and residency, she received postdoctoral training at the Institute and was appointed to faculty in 1996. Dr. Vuori was selected as a PEW Scholar in the Biomedical Sciences in 1997. She has been co-Director of the Conrad Prebys Center for Chemical Genomics, housed at Sanford Burnham Prebys, since its inception in 2005. She was appointed Deputy Director of the Institute’s NCI-Designated Cancer Center in 2003, and Director of the Cancer Center in 2006. In 2008, she was appointed Executive Vice President for Scientific Affairs at Sanford Burnham Prebys. She was President of the Institute from 2010 to 2022.
Related Disease
Brain Cancer, Breast Cancer, Cancer, Leukemia/Lymphoma, Lung Cancer, Ovarian Cancer, Prostate Cancer
Dr. Vuori’s research is aimed at unraveling the cell mechanisms of the most life-threatening aspect of cancer, which is cancer metastasis. Metastasis is responsible for nearly all deaths in cancer patients, and understanding of the mechanisms that turn a cancer from a locally growing tumor into highly metastatic cancer cells will provide clues how to prevent this step in cancer progression. All cells in our body stick to one another and to the packaging material, or extracellular matrix, around them. This adhesion is essential for cell survival; if cells become detached from their microenvironment, they will die through a process known as apoptosis. This phenomenon, which is called adhesion dependency of survival, is one of the safeguards that maintain the integrity and normal function of tissues, and prevent cells from becoming cancerous. Normal cells cannot detach from their tissue and establish themselves somewhere else, because they will die on the way. Yet cancer cells somehow get around this requirement; they trespass aggressively into other tissues and metastasize to distant sites in the body without dying. Dr. Vuori’s work is aimed at identifying the molecular mechanisms that in normal cells makes them adhesion-dependent; false action of the very same mechanisms is likely to be the key step in allowing cancer cells to metastasize.
Giovanni Paternostro earned his PhD in Biochemistry from the University of Oxford, England, in 1997. He has obtained his MD and Board Certification in Cardiology from the University of Rome, Italy. After postdoctoral training at the Imperial College School of Medicine, Hammersmith Hospital, London and at Sanford Burnham Prebys he was promoted to Research Investigator in 2001 and to Assistant Professor in 2003. In 2001 he was nominated member of the Whitaker Institute for Biomedical Engineering, UC San Diego. His research has been recognized by the 2002 Society for Geriatric Cardiology Basic Science Award and by the Ellison Medical Foundation New Scholar in Aging Award. Dr. Paternostro now holds adjunct faculty positions at Sanford Burnham Prebys Medical Discovery Institute and at the Department of Bioengineering, UC San Diego. His lab is located at Sanford Burnham Prebys.
Related Disease
Leukemia/Lymphoma, Ovarian Cancer
Our lab uses a systems biology approach to the study of complex diseases. Combined drug interventions are an increasingly common therapeutic approach to complex diseases, for example in cancer. Drugs are, however, usually developed individually and only later combined empirically in the clinic based on their known effects as single-therapy agents. We are interested in the problem of inducing selective cancer cell death. We have developed and validated search algorithms to discover optimal combinations of three or more drugs that would be infeasible to identify by fully combinatorial searches. In our procedure the optimization is not carried out in silico, but directly in an in vivo high-throughput system, where the response to therapeutic combinations is used as information to guide the system toward improved combinations using an iterative algorithm. System-wide molecular measurements (for example metabolomics and transcriptomics) and models can also be incorporated in these algorithms. It is useful to view the information processing by our experimental cellular systems as biological computations, since the algorithms we use are indeed often derived from algorithms that are implemented in silico in other scientific fields.
We also use the fruit fly (Drosophila) to study cardiac and metabolic alterations caused by aging and hypoxia, using high-throughput physiological measurements, NMR metabolomics and models of metabolism.
Our multi-disciplinary team is composed of biomedical and computational scientists, and we have close collaborations with physicists, engineers and bioengineers.
Nicholas Cosford, PhD has served on the Sanford Burnham Prebys Board of Trustees since 2023. He is the first faculty member to do so.
Cosford joined the Sanford Burnham Prebys faculty in 2008 as an associate professor. In 2013, he became a full professor. His lab investigates the interactions of small molecule compounds with therapeutically important proteins and cellular signaling pathways. With a specific focus on the discovery and optimization of compounds that might treat cancer, central nervous system diseases and infectious diseases.
Prior to joining Sanford Burnham Prebys in 2005, Cosford worked in both the biotechnology and pharmaceutical industries. At Sibia Neurosciences and at Merck Research Laboratories, he directed multidisciplinary research teams focused on small-molecule hit-to-lead optimization and was responsible for moving several lead compounds through to the clinical phase, including a nicotinic agonist (Altinicline) from research to Phase II clinical trials for treating Parkinson’s disease.
He is an author of more than 90 peer-reviewed, published scientific papers, and has been issued more than 40 issued patents, with an additional 40 patent applications pending.
Cosford has a Bachelor of Science degree in chemistry from the University of Bath in England and Doctor of Philosophy degree in organic chemistry from Emory University in Atlanta, GA.
Related Disease
Alzheimer’s Disease, Amyotrophic Lateral Sclerosis (Lou Gehrig’s Disease), Bone Mineralization Disorders, Breast Cancer, Cancer, Neurodegenerative and Neuromuscular Diseases, Neurological and Psychiatric Disorders, Ovarian Cancer, Pancreatic Cancer, Prostate Cancer
We are interested in investigating the interactions of small molecule compounds with therapeutically important proteins and cellular signaling pathways. One aspect of our research emphasizes the use of medicinal chemistry and chemical biology approaches to probe intracellular pathways that regulate cell survival and cell growth. Another area of active research is the development of synthetic chemistry methodology using microfluidic technology for the rapid synthesis of biologically active small molecules. Therapeutically, we are primarily focused on the discovery and optimization of compounds that have the potential to treat cancer, CNS diseases and infectious diseases.