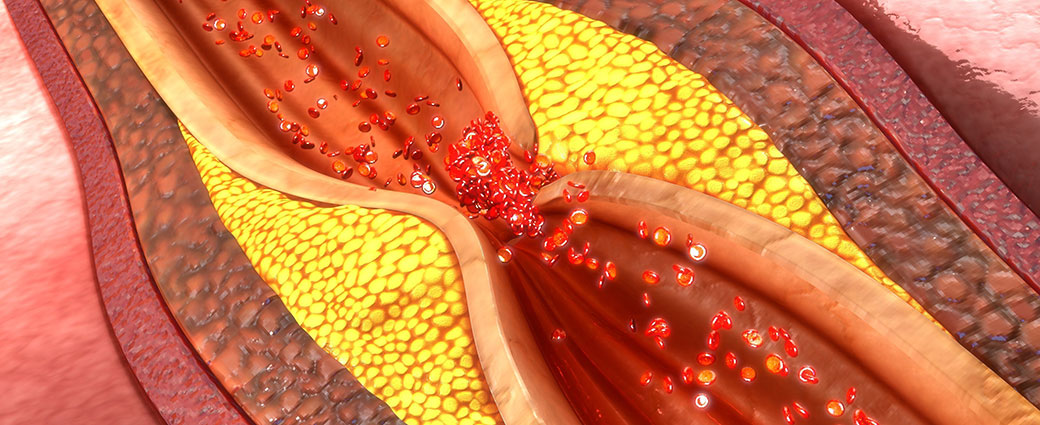
Atherosclerosis, the buildup of plaques inside arteries, is a key step in the development of cardiovascular disease, the leading cause of death in the United States. Elevated cholesterol levels are a risk factor for atherosclerosis, as the molecule is one of the building blocks of these plaques. However, since cholesterol is also essential in healthy cells, scientists are researching how cholesterol biology is controlled to better understand the changes that lead to disease.
Laszlo Nagy, MD, PhD, professor and director of the Genomic Control of Metabolism Program, recently collaborated with Peter Tontonoz, MD, PhD, professor, the leading senior scientist of the study and Francis and Albert Piansky Endowed Chair in Pathology and Laboratory Medicine at the UCLA David Geffen School of Medicine, to assess a network of molecules that control cholesterol transport out of macrophages, immune cells normally associated with inflammation.
“Although macrophages are usually thought of as the white blood cells that ingest invading bacteria and cleaning up cell debris after injury or infection, they can also enter a ‘alternatively activated’ state to help tissue repair and remodeling,” Nagy says. “In blood vessels, these repair state macrophages protect the body by removing cholesterol from the bloodstream. However, the accumulation of excess cholesterol in macrophages is a key event in the development of atherosclerosis. How macrophages control cholesterol transport is not well understood, but needs to be explored to better understand atherosclerosis.”
“We were interested in how macrophages are able to switch on the gene Abca1, the gene that encodes the protein that pumps cholesterol out of these cells,” Nagy explained. “We used our expertise in epigenomics to define regions of the genome that controlled the amount of Abca1 RNA produced.”
By examining long non-coding RNA strands that regulate gene expression, the study identified an RNA called MeXis that increases the expression of Abca1. Although MeXis cannot start transcription of Abca1 by itself, it does impact the ability of other proteins to transcribe the gene.
“Using our molecular tools, we were able to show that MeXis recruited another protein that helps to start transcription to Abca1, says Nagy. “Without MeXis, this protein did not interact with Abca1 and transcription was dramatically reduced, even when the cells received signals to start the process of ridding themselves of cholesterol.
“The more we understand about the biological processes that control cholesterol metabolism the better informed we are to develop strategies to prevent and treat atherosclerosis, says Nagy. “This study reveals key insights on the regulation of Abca1, which could ultimately lead to new therapeutic approaches.”
The study was published in Nature Medicine.