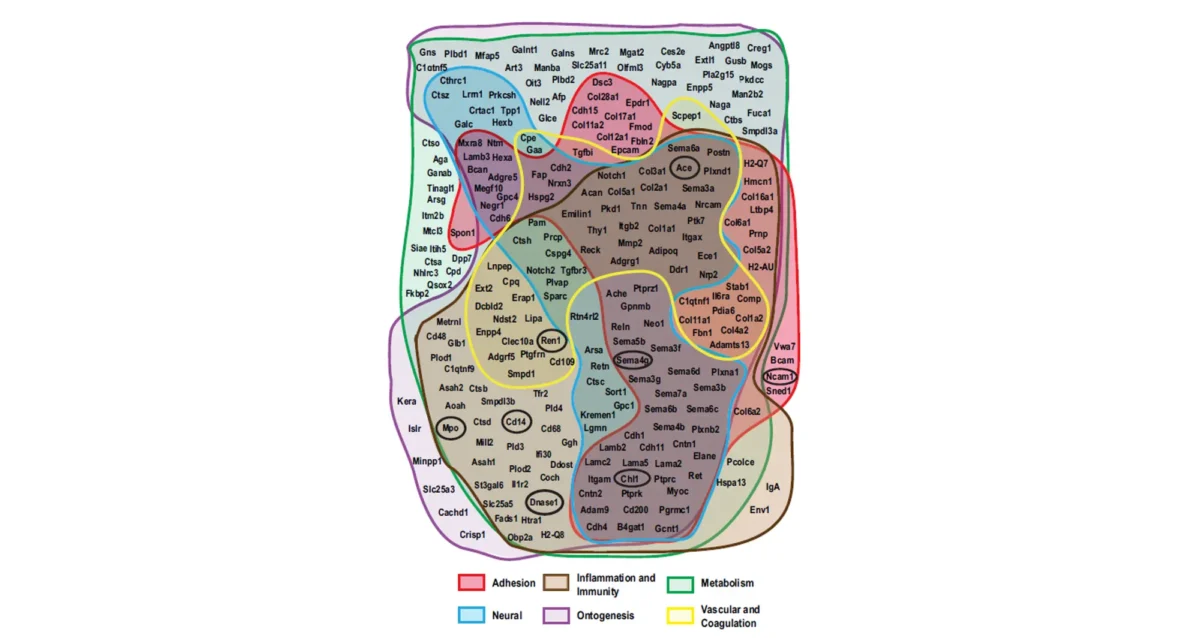
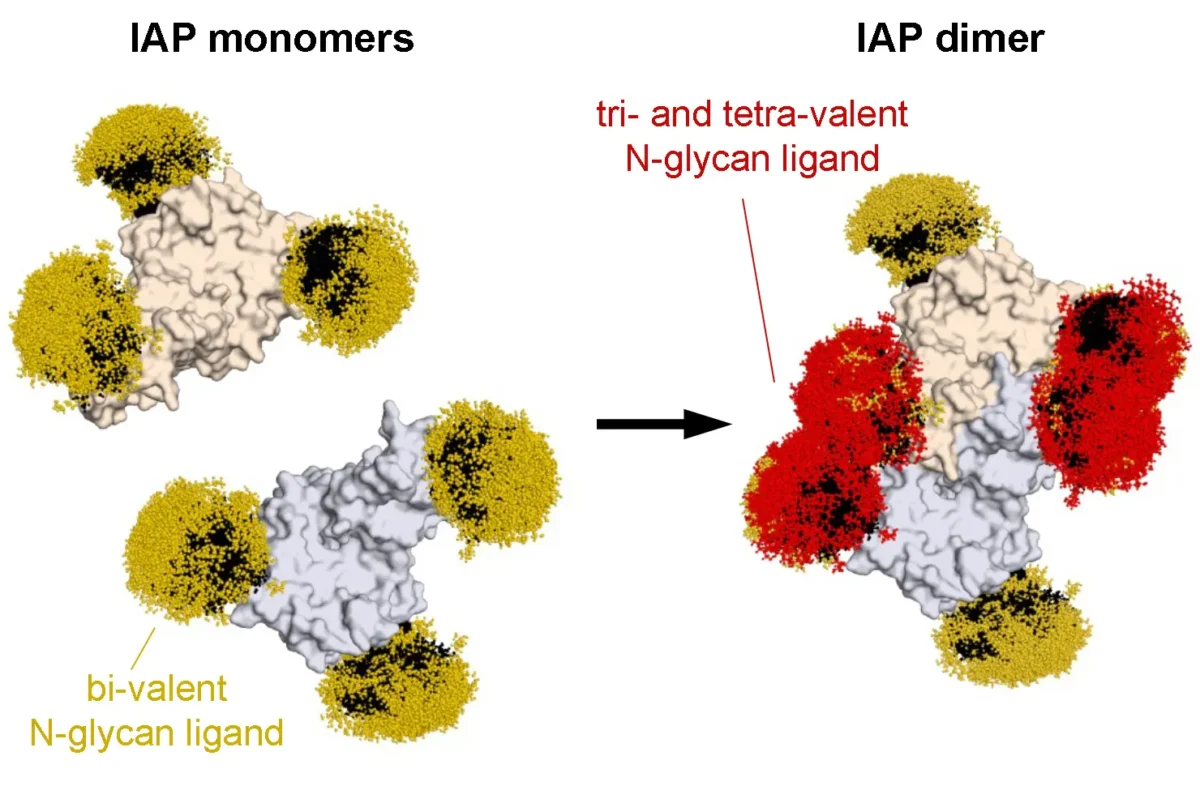
Sanjeev S. Ranade studies how transcription factors specifically control the development and function of cardiac cells — and what happens when things go wrong.
Transcription factors (TF) are proteins that initiate and regulate the transcription of genes, essentially turning genes on and off, boosting or repressing their activity. At last count, there were over 1,500 known TFs, but the contribution of most of the TFs to life and health is unknown.
In particular, Ranade focuses on how disrupted cell-to-cell signaling caused by mutations in TFs can cause congenital heart defects or CHDs.
“My research is focused on understanding why young children are born with heart defects. What are the principles and rules that allow our hearts to develop in the first place and why do these rules get broken in some cases? This is really important because nearly 1 in 100 children are born with some form of heart defect and many of these children will suffer from heart disease at much earlier stages in life compared to the general population.”
For his doctorate in molecular biology at Scripps Research in San Diego, Ranade studied ion channels — proteins that span cell membranes, allowing passage of ions or charged molecules from one side of the membrane to the other. The channels serve many critical functions, including transmitting signals involved in cell-cell communications and muscle contraction.
Working as a post-doctoral fellow and staff research scientist in the lab of Deepak Srivastava, M.D. at Gladstone Institutes, Ranade looked at how genetics and cell biology were connected and how disruptions to these connections led to children with heart defects.
The 24th Annual Biomedical Research Symposium brought together postdocs, graduate students, staff scientists, administrators, and faculty for a day that…
New and rapidly developing technologies, such as cryo-electron (cryo-EM) and artificial intelligence, are providing the tools to revolutionize biomedical research,…
Aug 2, 2023
Pier Lorenzo Puri earned his MD at the University of Rome “la Sapienza” in 1991. Dr. Puri completed his internship in Internal Medicine at the hospital “Policlinico Umberto I” (Rome) from 1992 to 1997, and defended an experimental thesis on the vascular effects of angiotensin II to graduate as Specialist in Internal medicine at the University of Rome “la Sapienza” in 1997. During this time he was frequently working at the Freien University of Berlin, as visiting scientist at the Deprtment of Biochemistry and Molecular Biology, to perform experiments of protein and DNA microinjection in cultured cells. Dr. Puri trained as a post-doctoral fellow at the University of California San Diego (UCSD), in the department of Cell Biology, under the supervision of Dr. Wang, from 1997 to 2001. He was appointed as Staff Scientist at the Salk Institute (La Jolla) in 2001, and became an Assistant Telethon Scientist at the Dulbecco Telethon Institute in Rome in 2002. He was upgraded to Associate Telethon Scientist at the Dulbecco Telethon Institute in Rome since 2007 and became Senior Telethon Scientist, Dulbecco Telethon Institute, in 2012, but declined this position. Dr. Puri joined Sanford Burnham Prebys as an Assistant Professor in 2004. He has been promoted to Associate Professor in 2010 and full Professor in 2015. From 2008 to 2016 Dr. Puri served as Adjunct Professor of Pediatrics at the University of California, San Diego. From 2008 to 2013 Dr Puri was an Associate Member of Sanford Children’s Health Research Center. Dr Puri has been Director of the laboratory of Epigenetics and Regeneration at Fondazione S. Lucia, Roma, Italy, but stepped down this position since 2019.
Education
University of California San Diego, Postdoctoral, Department of Biology University of Rome La Sapienza, PhD, Internal Medicine University of Rome La Sapienza, MD, Internal Medicine University of Rome La Sapienza, Undergraduate, Internal Medicine
Other Appointments
2020-2024: Member of the Science Advisory Board (SAB) European Commission-funded Consortium BIND (Brain Involvement In Dystrophinopathies) 2015-2019: Standing Member, NIH Study Section (SMEP) 2010-present: Member of Editorial Board of Skeletal Muscle
Phenomena or Processes
Adult/Multipotent Stem Cells, Aging, Cell Biology, Cell Cycle Progression, Cell Differentiation, Cell Signaling, Cellular Senescence, Development and Differentiation, Disease Therapies, DNA Damage Checkpoint Function, Epigenetics, Gene Regulation, Phosphorylation, Regenerative Biology, Signal Transduction, Transcriptional Regulation
Anatomical Systems and Sites
General Cell Biology, Musculoskeletal System
Research Models
Clinical and Transitional Research, Cultured Cell Lines, Human Adult/Somatic Stem Cells, Mouse Embryonic Stem Cells, Mouse Somatic Stem Cells, Primary Human Cells
Techniques and Technologies
Bioinformatics, Cellular and Molecular Imaging, Gene Expression, Genomics
Puri’s lab group investigates the molecular and epigenetic regulation of gene expression in skeletal muscle progenitors and other muscle-resident cell types (including fibro-adipogenic progenitors, cells from the inflammatory infiltrate, cellular components of neuro-muscular junctions) during physiological and pathological perturbations of skeletal muscle homeostasis.
We use molecular, biochemical and epigenetic tools to understand structural and functional principles of the 3D genome organization that regulates gene expression during muscle regeneration and diseases.
A topic of particular interest is the analysis of chromatin interactions that define the 3D genome organization and the identification of structural and functional interactions that regulate cell type-specific patterns of gene expression in response to cues released within the skeletal muscle regenerative environment in health and disease conditions, such as muscular dystrophies and other neuromuscular diseases.
The knowledge derived from our studies is instrumental to elucidate the pathogenesis of muscular disorders and discover pharmacological interventions that promote muscle regeneration to repair diseased muscles.
Current translational focus is devoted to:
the study of the therapeutic potential of HDAC inhibitors for treatment of Duchenne Muscular Dystrophy (DMD)
the identification of genome variants associated to DMD patient-specific patterns of expression of disease-modifier genes that can account for individual trends of disease progression beyond the common genetic deficiency of dystrophin
the effect of dystrophin deficiency and restoration by gene therapy on 3D genome and transcriptional output of DMD myofibers; the therapeutic potential of extracellular vesicles released by fibro-adipogenic progenitors of DMD skeletal muscles exposed to HDACi.
Puri Lab
Pier Lorenzo Puri’s Research Report
Puri’s lab group investigates the molecular and epigenetic regulation of gene expression in skeletal muscle progenitors and other muscle-resident cell types (including fibro-adipogenic progenitors, cells from the inflammatory infiltrate, cellular components of neuro-muscular junctions) during physiological and pathological perturbations of skeletal muscle homeostasis.
We use molecular, biochemical and epigenetic tools to understand structural and functional principles of the 3D genome organization that regulates gene expression during muscle regeneration and diseases
A topic of particular interest is the analysis of chromatin interactions that define the 3D genome organization and the identification of structural and functional interactions that regulate cell type-specific patterns of gene expression in response to cues released within the skeletal muscle regenerative environment in health and disease conditions, such as muscular dystrophies and other neuromuscular diseases.
The knowledge derived from our studies is instrumental to elucidate the pathogenesis of muscular disorders and discover pharmacological interventions that promote muscle regeneration to repair diseased muscles
Current translational focus is devoted to:
the study of the therapeutic potential of HDAC inhibitors for treatment of Duchenne Muscular Dystrophy (DMD)
the identification of genome variants associated to DMD patient-specific patterns of expression of disease-modifier genes that can account for individual trends of disease progression beyond the common genetic deficiency of dystrophin
the effect of dystrophin deficiency and restoration by gene therapy on 3D genome and transcriptional output of DMD myofibers; the therapeutic potential of extracellular vesicles released by fibro-adipogenic progenitors of DMD skeletal muscles exposed to HDACi.
1. Epigenetic regulation of skeletal myogenesis by histone acetyltransferases and deacetylases
Our earlier identification and characterization of acetyltransferases p300/CBP and PCAF, as transcriptional co-activators, and the histone deacetylases HDACs, as transcriptional co-repressors, of the myogenic determination factor MyoD1-3, respectively, inspired the experimental rationale toward exploiting pharmacological inhibition of HDAC to promote skeletal myogenesis.
2. HDAC inhibitors as pharmacological intervention in DMD and other muscular dystrophies
Puri lab discovered that dystrophin-activated nNOS signalling controls HDAC2 activity, thereby revealing a previously unrecognized link between constitutive activation of HDAC2 and alteration of the epigenetic landscape of dystrophin-deficient muscles6,7. This discovery established the rationale for using HDAC inhibitors to counter the progression of Duchenne muscular dystrophy (DMD), by correcting aberrant HDAC activity in dystrophin-deficient muscles8-11.
3. Control of chromatin structure in muscle cells by regeneration-induced signaling pathways
Upon the discovery and characterization of intracellular signaling pathways (i.e. p38, ERK and AKT cascades) that regulate muscle gene expression in myoblasts, in earlier studies during Puri’s postdoctoral training, Puri lab has revealed the mechanism by which muscle environmental cues are converted into epigenetic changes that regulate gene expression in healthy and diseased muscles, via extracellular signal-activated kinase targeting of chromatin-modifying enzymes. These studies provided the first evidence that regeneration activated p38 and AKT signaling cooperatively direct assembly and activation of histone acetyltransferases and chromatin remodeling SWI/SNF complex at myogenic loci in muscle progenitors12,13,15. Moreover, we discovered that regeneration-activated p38 targets Polycomb Repressory Complex (PCR2) at Pax7 locus to promote formation of repressive chromatin during satellite cells a ctivation14.
4. Epigenetic basis for activation of the myogenic program in ESCs and other pluripotent cell types
Puri lab studied the epigenetic determinants of human embryonic stem cells (hESCs) and induced pluripotent stem cells (hiPSCs) commitment to skeletal myogenesis, by investigating the hESC resistance to direct conversion into skeletal muscle upon ectopic expression of MyoD, which can otherwise reprogram somatic cells into the skeletal muscle lineage. These studies showed that hESC and hiPSC resistance to myogenic conversion is caused by the lack of expression of one structural component of the SWI/SNF chromatin remodelling complex – BAF60C – which is specifically induced in embryoid bodies13. Based on these studies, we have recently established a protocol of hESC-derived 3D contractile myospheres that offers the unprecedented opportunity to dissect and analyze the epigenetic dynamics that underlie the formation of skeletal muscles and to identify changes in the epigenome induced by contractile activity in healthy vs dystrophin-deficient myofibers16,20. We have also determined the identity of the general transcription factors implicated in the activation of skeletal myogenesis17, and we have discovered that replicative senescence is associated with acquisition of resistance to MYOD-mediated activation of muscle gene expression, caused by the constitutive activation of DNA damage repair (DDR) response that impairs cell cycle progression and MYOD activity18. Finally, our recent work has elucidated the mechanism by which MYOD regulates high-order chromatin interactions to define the tri-dimensional (3D) nuclear architecture for the activation of skeletal myogenesis during human somatic cell reprogramming into skeletal muscles19.
5. Identification, functional, phenotypic and molecular characterization of muscle-interstitial cells – (the fibroadipogenic progenitors – FAPs) in healthy and diseased muscles.
Our work has elucidated the molecular determinants of the interplay between adult muscle stem cells and cellular components of their functional niche (i.e. FAPs), by identifying regulatory networks implicated in compensatory or pathogenic regeneration, and suggesting “disease stage-specific” responses to pharmacological treatment of neuromuscular disorders, such as DMD. Indeed, we have shown that HDACi promote compensatory regeneration and prevent fibro-adipogenic degeneration in mdx mice at early stages of diseases, by targeting a population of muscle interstitial cells – FAPs8 – and have identified a HDAC-regulated network that controls expression of myomiRs and alternative incorporation of BAF60 variants into SWI/SNF complexes to direct the pro-myogenic or fibro-adipogenic FAP activity21. Furthermore, we have recently identified specific subpopulations of FAPs (subFAPs) in physiological conditions and disease22 and we have discovered that specific subFAPs expand and adopt pathogenic phenotypes upon muscle denervation23 or in muscles of patients affected by type 2 diabetes24.
Elena Pasquale earned her PhD in biology from the University of Parma, Italy. She did postdoctoral work at Cornell University, after which she was appointed Research Assistant Professor at University of Parma. Following a second postdoctoral training period at the University of California in San Diego, Dr. Pasquale was appointed Assistant Research Biologist at that institution. Dr. Pasquale was recruited to Sanford Burnham Prebys in 1990.
Related Disease
Cancer, Neurodegenerative and Neuromuscular Diseases, Skin Cancer and Melanoma
Cancer, Neurodegenerative and Neuromuscular Diseases
Receptor tyrosine kinases of the Eph family and their ligands, the ephrins, represent an important cell communication system that controls a vast array physiological and disease processes. For example, Eph receptors and ephrins take part in the formation of blood vessels, including the blood vessels that feed tumors, and regulate the malignant properties of cancer cells and their interplay with the tumor microenvironment. They also regulate the formation, plasticity and regeneration of neuronal circuits as well as neurodegenerative processes such as those occurring in amyotrophic lateral sclerosis (ALS) and Alzheimer’s disease. The signal transduction mechanisms of Eph receptors are intriguing, and complex, because these receptors engage in multiple modes of signaling. Binding to ephrin ligands on the surface of neighboring cells induces canonical signaling involving receptor clustering, autophosphorylation on tyrosine residues, and kinase activity-dependent downstream signaling. Binding to the Eph receptors can also cause the ephrins, which have a cytoplasmic domain or a GPI-anchor, to transmit signals. This leads to bidirectional signals emanating from Eph receptor-ephrin complexes positioned at sites of cell-cell contact. In addition, at least some Eph receptors can also signal through non-canonical mechanisms that are independent of ligand binding and kinase activity, for example through interplay with other receptor tyrosine kinase families and with serine/threonine kinases.
Our research investigates Eph receptor signaling activities in order to understand their role in normal physiology and in pathological conditions such as cancer and neurodegenerative disorders. This knowledge is useful for the development of disease treatments based on modulating Eph receptor/ephrin activities. Ongoing efforts in our laboratory also focus on the development of agents targeting Eph receptors for research and translational applications.
Elena Pasquale’s Research Report
We discovered several Eph receptors and ephrins, and research in our laboratory is dedicated to the characterization of Eph receptor signal transduction mechanisms and biological functions using biochemical, mass spectrometry, molecular biology and cell biology approaches in conjunction with animal models. We have identified tyrosine and serine/threonine phosphorylation sites of Eph receptors and ephrins using mass spectrometry and investigated the signaling role of these phosphorylation sites. For example, our past work showed that two conserved tyrosine phosphorylation sites in the juxtamembrane segment of the Eph receptors not only mediate association with binding partners but also regulate receptor kinase activity. We also found that the SRC and ABL non-receptor tyrosine kinases and the SHEP1 scaffolding protein are binding partners of the Eph receptors, and we identified signaling connections between Eph receptors and integrins. We also found that EphA4 is highly expressed in the adult brain, where it regulates synaptic connections. More recent work in our laboratory focuses on elucidating signaling pathways that mediate the activities of Eph receptors in cancer cells.
Tumor Suppression and Tumor Promotion by Eph Receptors
Many Eph receptors are highly expressed in tumors, but their role in cancer is incompletely understood and likely depends on the cellular context. Certain Eph receptors and ephrins promote tumor angiogenesis. We showed that the EphA2 receptor is upregulated in the tumor vasculature together with the ephrin-A1 ligand, which suggested a role in tumor angiogenesis that is now well established. We also found that the EphB4 receptor expressed on the surface of breast cancer cells can promote tumor xenograft growth by enhancing blood vessel formation through interactions with its preferred ligand, ephrin-B2, present in tumor endothelial cells. Additional intriguing roles for the Eph receptors in cancer progression have also emerged. We found that canonical signaling by the EphB4 receptor is low in breast cancer cells and that ephrin-induced stimulation of EphB4 kinase activity inhibits breast cancer cell malignancy in culture and tumor growth in vivo (Figure 1A) through inhibition of the CRK proto-oncogene. More recently, we elucidated an additional mechanism of tumor suppression mediated by canonical ephrin-induced EphA2 signaling (Figure 1A), which leads to inhibition of the AKT-mTORC1 oncogenic pathway through interplay of EphA2 with a phosphatase that dephosphorylates the AKT serine/threonine kinase.
Figure 1. Dual activities of Eph Receptors in Cancer Cells. (A) Eph receptor-ephrin binding at cell-cell contact sites results in the dimerization/clustering of Eph receptor-ephrin complexes, and initiation of canonical signals through the receptor cytoplasmic domain. Signals through the ephrins can also be generated. Tyrosine phosphorylation sites (yellow circles) promote Eph kinase activity and also provide binding sites for signaling proteins containing SH2 domains. Other effectors also mediate Eph signals, including PDZ domain-containing proteins. The Eph receptor domains are indicated on the left; LBD, ligand-binding domain. (B) Eph receptors can potentiate the oncogenic effects of other receptors. These activities are independent of ephrin binding and/or kinase activity and their mechanism is not well understood but in some cases depends on Eph receptor phosphorylation on serine/threonine residues (red circle).
There is also evidence that some Eph receptors can increase cancer cell malignancy through non-canonical ephrin-independent and/or kinase-independent signaling activities (Figure 1B), which is the subject of ongoing work. These tumor promoting activities include inducing invasiveness and metastasis, epithelial-to-mesenchymal transition, stem cell-like features and drug resistance.
Eph Receptor Mutations in Cancer
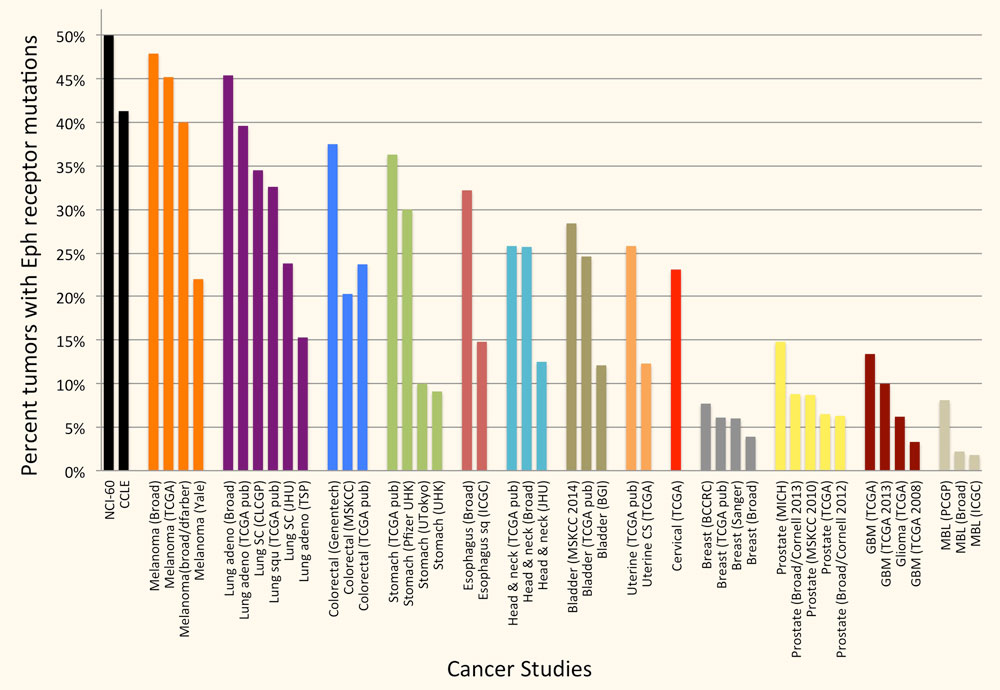
The Eph receptors are frequently mutated in many types of cancer. In particular, genome-wide sequencing studies have detected somatic mutations in one or more Eph receptors in 25%-45% of melanomas, 15%-45% of lung cancers, 25-40% of colorectal cancers and 12%-25% of head and neck and uterine cancers (Figure 2), but very limited information is available on the effects of the mutations. Studies by ours and other groups have shown that a number of EphA2 and EphA3 mutations inactivate Eph receptor canonical signaling by disrupting ephrin binding or kinase activity, consistent with a role of canonical signaling in tumor suppression. Ongoing work in our laboratory focuses on characterizing the functional effects of Eph receptor mutations in cancers such as melanoma, and investigating whether the mutations shift the balance of the Eph receptor signaling activities from tumor suppression to tumor promotion. We are also interested in the interplay of Eph receptor mutations with mutations affecting well-established oncogenes and tumor suppressor genes. Understanding the effects of Eph receptor mutations in cancer cells will help shed light on the role of the Eph receptor/ephrin system in cancer cell transformation, malignant progression and drug resistance.
Figure 2. A large percentage of tumor specimens and cell lines harbor one or more Eph receptor mutations. Groups of bars of the same color represent studies of the same cancer type. The cancers with most Eph receptor mutations are shown; other tumor types have fewer or no Eph receptor mutations. The graph is based on data from cBioPortal for Cancer Genomics (www.cbioportal.org).
Peptides Targeting Eph Receptors
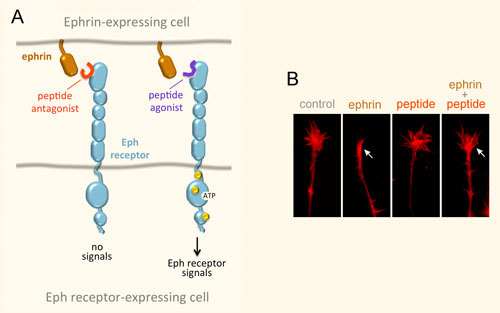
We have identified a number of peptides that bind to Eph receptors and inhibit ephrin binding by using phage display approaches. Collaborating groups have elucidated the structural features of several of these peptides in complex with the ligand-binding domain of Eph receptors, demonstrating that the peptides bind to the ephrin-binding pocket in the ligand-binding domain (Figure 3A). Most of the peptides are antagonists, but the peptides targeting EphA2 are agonists that activate receptor signaling and endocytosis similarly to the natural ephrin ligands. Interestingly, some of the identified peptides are highly specific and bind to only one Eph receptor family member. This is unlike the natural ephrin ligands, each of which promiscuously binds to multiple Eph receptors. Thus, Eph receptor-targeting peptides represent valuable pharmacological tools to study the functional importance of specific Eph receptors in tumors and the nervous system. Furthermore, they could be used as leads to develop therapies against cancer and neurological disorders, and to promote neural repair after nervous system injury (Figure 3B). Finally, our peptides have been used by other groups to deliver conjugated imaging agents, drugs and nanoparticles to Eph receptor-positive tumors. Current work focuses on identifying novel Eph receptor-targeting agents (such as peptides and small molecules) as well as improving the existing ones in collaboration with medicinal chemists and structural biologists, and evaluating them in cell culture and in vivo animal models.
Figure 3. Peptides can target the ephrin-binding pocket of Eph receptors with high affinity and specificity, affecting receptor function. (A) Peptides targeting the Eph receptors can function as antagonists that inhibit ephrin binding and receptor signaling, or in some cases as agonists that mimic the ephrins by activating Eph receptor signaling. Yellow circles indicate tyrosine phosphorylation sites in the activated Eph receptor. (B) An EphA4 peptide antagonist blocks ephrin-induced growth cone collapse in EphA4-expressing axons, suggesting its usefulness for promoting neural repair. The arrow in the second panel marks a growth cone collapsed due to ephrin treatment; the arrow in the fourth panel marks a growth cone that did not collapse following ephrin treatment in the presence of a peptide antagonist.
Andrei Osterman is a Professor in the Immunity and Pathogenesis Program Program at the Infectious and Inflammatory Disease Center of Sanford Burnham Prebys (since August 2003). He received his doctorate from Moscow State University in 1983, did postdoctoral work UT Southwestern Medical Center, and held the position of the Director and then Vice President of Research at Integrated Genomics in 1999-2003. Dr. Osterman is one of the founders of the Fellowship for Interpretation of Genomes (FIG), a nonprofit research organization that launched the Project to Annotate 1,000 Genomes in 2003. FIG provides the open-source integration of all publicly available genomes and tools for their comparative analysis, annotation, and metabolic reconstruction.
Related Disease
Breast Cancer, Cancer, Infectious Diseases, Radiation Damage, Skin Cancer and Melanoma
The main focus of Dr. Osterman’s research team is on fundamental and applied aspects of the key metabolic subsystems in a variety of species, from bacteria to human. This group uses a systems biology approach to reconstruct and explore metabolic and transcriptional regulatory networks. This approach combines comparative genomics and other bioinformatic techniques with biochemical and genetic experiments for pathway, gene and target discovery. Using this approach this group predicted and experimentally verified numerous enzyme families in the metabolism of cofactors, carbohydrates, and amino acids. Recent breakthroughs included prediction and characterization of novel transporters, transcriptional regulators and carbohydrate utilization pathways in a number of model bacterial systems. Applications in the field of infectious disease include identification of novel drug targets and structure-based development of novel anti-infective agents. New directions in cancer research are based on application of metabolic profiling technology for identification of novel diagnostic and therapeutic targets. Other directions of the on-going research include bioinformatics of regulatory proteolysis and applications of structural modeling for exploration of metabolic networks and gene discovery.
Sanford Burnham Prebys scientists say that understanding the potential pitfalls of using artificial intelligence and computational biology techniques in biomedical…
After a successful teaching career at the University of Michigan I have had the privilege to “reboot” my research career at Sanford Burnham Prebys where I have had the opportunity to develop novel methodologies to understand cardiomyopathy. I have also had the opportunity to work with NASA scientists to do experiments on the International Space Station.
Education
Postdoctoral Fellow, Stanford University, Palo Alto, CA, Neurochemistry Postdoctoral Fellow, University of Texas Medical School, Houston, TX, Neuroscience NIMH PhD, Wesleyan University, Middletown, CT, Neuroscience NIMH B.A., Lehigh University, Bethlehem, PA, Biology
Prestigious Runding Awards or Major Collaborative Grants
2015-2020: NIH R01 HL132241-01A1 – Using Drosophila genetics to identify molecular links between ion channel dysfunction and pathological cardiac remodeling. (PI) 2013-2018 NASA NRA #NNH12ZTT001N – The effects of microgravity on cardiac function, structure and gene expression using the Drosophila model. (Co-I)
Honor and Awards
2014: Space Florida International Space Station Research Competition Winner – Co-investigator – One of three Basic Research proposals selected for launch aboard SpaceX3 – Mission completed, live flies returned on May 18,2014 2001: Excellence in Teaching Award, University of Michigan 1997: Excellence in Teaching Award, University of Michigan 1986-1988: National Institute of Mental Health Fellowship 1983-1985: National Institute of Mental Health Fellowship 1981: Sigma Xi Research Award 1980 MBL Scholarship, Neural Systems and Behavior Course 1971-1975: National Merit Scholarship, Lehigh University
Board Appointments
2018-present: Board member American Society for Gravitational and Space Research
Anatomical Systems and Sites
Cardiovascular System, Heart
Research Models
Drosophila, Larval Zebrafish Heart, Zebrafish
Techniques and Technologies
Biophysiology, Cellular and Molecular Imaging, Fluorescence Microscopy, Gene Silencing, Genetics, In vivo Modeling, Ion Channels, Live Imaging, Microarrays, Microscopy and Imaging, Molecular Genetics, RNA Interference (RNAi), Semi-automated Optical Heartbeat Analysis (SOHA), Systems Biology, Transgenic Organisms
The Ocorr Lab is investigating the cellular and molecular basis of adult heart function and cardiomyopathies using the genetic model system Drosophila.
We use functional, electrophysiological, biochemical and immunohistochemical techniques that allow us to examine the roles of genes and gene products in cardiac channelopathies and stress-related cardiomyopathies.
Our lab pioneered the development of a novel methodology (Semi-automatic Optical Heartbeat Analysis, SOHA) that permits the quantification of heartbeat parameters in model systems with small hearts.
Using this system we have identified several ion channels in the fly heart that play prominent roles in repolarization of the human heart and cause arrhythmia in both the fly and in humans when mutated. We also have developed a number of other disease models including a diabetic-like cardiomyopathy induced by high sugar diet and hypoxia-induced cardiomyopathy.
Recently we have begun collaborations with NASA (by winning a Space Florida International Space Station Research Competition). We are using the fly to uncover the molecular/cellular basis for cardiac and muscle atrophy in astronauts exposed to extended periods of microgravity despite extensive exercise regimes aboard the ISS. Our flies were launched aboard SpaceX 3 for a month-long exposure to micro-gravity.
Karen Ocorr’s Research Report
My lab is working to understand the cellular and molecular basis of heart disease. One project is focused on the genetic basis of Atrial Fibrillation. This project is a collaborative one with the lab of Alexandre Colas. We are combining two model systems, the fly in my lab and human induced cardiomyocytes in his lab, to identify AFib genes that have been implicated from patient studies. Another project focuses on the role of metabolism in cardiomyopathies. This is because obesity and metabolic syndrome are linked to an increased risk of heart disease. We are studying the role of a key metabolic signaling molecule in hypertrophic cardiomyopathy. A separate effort is focused on the role of gravity in heart function. These studies will provide important information for future habitants of colonies on the moon and Mars. But they are also relevant to patients who are bedridden and to patients with muscle wasting (sarcopenia).
After receiving his early training in clinical chemistry/biochemistry at the University of Buenos Aires, Argentina, Dr. Millán first joined the La Jolla Cancer Research Foundation (LJCRF) in 1977, the predecessor of Sanford Burnham Prebys, as a trainee in clinical enzymology. He completed his PhD studies in Medical Biochemistry at the University of Umeå, Sweden and after post-doctoral stints in Copenhagen and LJCRF he was appointed to the faculty at SBP in 1986. He served as Professor of Medical Genetics in the Department of Medical Biosciences at his alma mater, Umeå University, Sweden, from 1995-2000. He was appointed Sanford Investigator at the Sanford Children’s Health Research Center at Sanford Burnham Prebys in 2008.
Honors and Recognition
2018: ASBMR Lawrence G. Raisz Award for Pre-clinical Research. 2001: Gold Medal of the Royal Academy of Medicine and Surgery, Murcia, Spain 1992: Honorary title of AcadémicoCorresponsal at the Royal Academy of Medicine and Surgery, Murcia, Spain.
Related Disease
Arthritis, Bone Mineralization Disorders, Cardiovascular Diseases, Colorectal Cancer, Crohn’s Disease (Colitis), Heart Disease, Inherited Disorders, Metabolic Syndrome, Peripheral Vascular Disease, Testicular Cancer
Phenomena or Processes
Cardiovascular Biology, Disease Therapies, Extracellular Matrix, Protein Structure-Function Relationships
Anatomical Systems and Sites
Cardiovascular System, Musculoskeletal System, Vasculature
Research Models
Mouse
The Millán laboratory works on understanding the mechanisms that control normal skeletal and dental mineralization and elucidating the pathophysiological abnormalities that lead to heritable soft bones conditions such as Hypophosphatasia (HPP) and to soft-tissue calcification, including vascular calcification, that is a hallmark in patients affected by a variety of rare genetic diseases as well as in chronic kidney disease. Dr. Millán’s research has already contributed to the implementation of a novel therapy for HPP, a genetic disease caused by deficiency in tissue-nonspecific alkaline phosphatase (TNAP) function, that leads to accumulation in the extracellular space of inorganic pyrophosphate (PPi), a potent inhibitor of mineralization. HPP is characterized by defective mineralization of bones (rickets or osteomalacia), and teeth that display a lack of acellular cementum, hypomineralized dentin and enamel, and periodontal defects. Dr. Millán’s team has demonstrated the effectiveness of enzyme replacement therapy using mineral-targeted recombinant TNAP (asfotase alfa) to prevent the skeletal and dental defects in the TNAP knockout mouse model of infantile HPP. This therapy was approved in 2015 for the treatment of patients with pediatric-onset HPP.
Current efforts, in collaboration with Professor Miyake’s group in Japan (https://www.nms-gt.org/en/members), focus on developing gene therapy as an alternative approach to treat HPP. Dr. Millán’s group has also identified key pathophysiological changes that lead to calcification of the arteries in animal models of generalized arterial calcification of infancy, pseudoxanthoma elasticum and related genetic diseases as well as in animal models of chronic kidney disease. His group, in collaboration with scientists at the Conrad Prebys Center for Chemical Genomics at Sanford Burnham Prebys, has developed proprietary compounds able to ameliorate the soft-tissue calcification in these conditions and clinical trials are now underway using these first-in-class small molecule inhibitors.
Jamey Marth is a Professor at Sanford Burnham Prebys. Dr. Marth’s previous positions included Professor of Medical Genetics at the Biomedical Research Centre, University of British Columbia; Investigator of the Howard Hughes Medical Institute and Professor of Cellular and Molecular Medicine at the University of California San Diego; and Professor and Director of the Center for Nanomedicine at the University of California Santa Barbara. Dr. Marth received a PhD degree in Pharmacology from the University of Washington where he trained in the laboratories of Roger M. Perlmutter, MD, PhD, and Nobel-laureate Edwin G. Krebs, MD.
Education
1987: PhD, University of Washington, Pharmacology 1984: BSc, University of Oregon, Genetics and Chemistry
Honors and Recognition
2024: Distinguished Service Award, Society for Glycobiology 2017: Karl Meyer Award, Society for Glycobiology 2009-2020: John Carbon Chair in Biochemistry and Molecular Biology 2009-2019: Duncan and Suzanne Mellichamp Chair in Systems Biology 2009: Julius Stone Lectureship Award: Society for Investigative Dermatology 1995-2009: Investigator Award, Howard Hughes Medical Institute 1991-1995: Faculty Scholarship, The Medical Research Council of Canada
Related Disease
Cancer, Colitis, Diabetes – General, Inflammatory/Autoimmune Disease, Sepsis
Dr. Marth is a molecular and cellular biologist specializing in diseases attributable to protein glycosylation. His education and training span molecular genetics, biochemistry, pharmacology, cell biology, immunology, hematology, developmental biology, microbiology, and glycobiology.
As an enzymatic process essential to cells, glycosylation produces saccharides linked by glycosidic bonds to proteins, lipids, and themselves, termed glycans. The vast majority of secreted and cell surface proteins are post-translationally modified by glycosylation during transit through the secretory pathway, termed glycoproteins. A widely used college level cell biology textbook authored by others denotes glycans as one of the four main families of the organic molecules of all cells, with lipids, proteins, and nucleic acids. Together they compose the macromolecules and other assemblies of the cell. The structures of glycans (and lipids) are, however, synthesized by template-independent processes, rendering them hard to predict and study. However, cells produce and regulate an abundant and diverse glycome of glycosidic linkages in which some of the biological information is decoded by one or more glycan-binding receptors, termed lectins.
Glycans and lectins represent a significant percentage of genes in the genomes of organisms, with several hundred present in mammals. Because glycan biosynthesis, diversification, and degradation rely upon corresponding gene and enzyme function, glycan function can be investigated similarly to other enzymatic and metabolic pathways, such as protein phosphorylation. In contrast however, studies of intact organisms are typically required to uncover the functions of protein glycosylation in mammals. Dr. Marth’s laboratory identified this model system requirement and has further focused on discovering how glycosidic linkages regulate proteins modified by N- and O-glycans and contribute to the molecular origins of common diseases and syndromes including colitis, diabetes, autoimmune disease, and sepsis.
To understand the nature and extent of the information within glycosidic linkages, the Marth Laboratory has applied multiple molecular approaches to investigate protein glycosylation in mice and humans. This includes the development of enabling technologies with broad applicability, such as conditional mutagenesis by Cre-lox recombination in living animals to determine gene function with temporal and spatial selectivity. His laboratory also develops and studies disease models that better represent real-world models of environmental factors that trigger common acquired human diseases. The laboratory is based on interdisciplinary research focused upon physiology and disease process regulated by protein glycosylation.
The physiological systems regulated by protein glycosylation are broad even when comparing among sequential biosynthetic steps, and our findings continue to indicate the presence of undiscovered information of medical relevance residing in the glycan linkages of glycoproteins.
Dr. Kumsta earned her degree as a Diplom Biologist/M.Sc. and PhD from the Technical University of Munich, Germany and performed her thesis research in the laboratory of Dr. Ursula Jakob at the University of Michigan. Dr. Kumsta joined Sanford Burnham Prebys and the lab of Malene Hansen as a postdoctoral fellow in 2009. In 2018 Caroline was promoted to Research Assistant Professor and then to Assistant Professor in 2021. Dr. Kumsta’s research focuses on the role of autophagy in hormetic stress responses, aging, and neurodegeneration in C. elegans and human tissues.
Education and Training
2018-2021: Research Assistant Professor, Sanford Burnham Prebys 2016-2018: Staff Scientist, , Sanford Burnham Prebys 2009-2016: Postdoctoral Fellow with Dr. Malene Hansen, Sanford Burnham Prebys 2009: Postdoctoral Associate with Dr. Ursula Jakob, Department of Molecular, Cellular and Developmental Biology, University of Michigan, Ann Arbor, USA 2005-2008: Doctor rerum naturalium (PhD) with Dr. Ursula Jakob, University of Michigan and Technical University of Munich 1999-2005: Diplom-Biologin Univ. (MS), Technical University of Munich, Germany
Honors and Awards
2024: R01 NIA:Hormetic regulation of autophagy in aging 2024: Selected as Member of Council of International Rising Stars (COIRS) of The Autophagy, Inflammation and Metabolism Center of Biomedical Research (NIH-funded AIM Center, University of New Mexico) 2023: P30 NIA: Pilot Grant, San Diego – Nathan Shock Center: Heterogeneity of autophagy during aging 2022: W.O.W. Award – For Wonderful Original Work, presented at the Annual Retreat of the Sanford Burnham Prebys Faculty 2022: NIA Fellow Award at 2022 Autophagy Gordon Research Conference 2015: Best Oral Presentation Prize, EMBO Workshop: The Regulation of Aging and Proteostasis 2013: AFAR Postdoctoral Fellowship: Transcriptional regulation of autophagy in promoting proteostasis upon hormetic stress 2013: Best Oral Presentation at the 12th Annual Poster Symposium at Sanford Burnham Prebys Medical Discovery Institute 2011: Fishman Fund Career Development Award 2011: AACR Postdoctoral Fellowship: Translational Control of Tumor Formation in C. elegans
Related Disease
Aging-Related Diseases
Phenomena or Processes
Autophagy
Research Models
C. elegans
“We are aging—not just as individuals but as a world. In 2020, about 727 million people worldwide were 65 and older. By 2050, that total is projected to increase to 1.5 billion — 1 in every 6 of the earth’s inhabitants.” – Global Aging, National Institute on Aging
As we age, we face an increased risk of developing age-related disorders, such as neurodegenerative diseases, because of the increased cellular accumulation of damaged biomolecules, including protein aggregates, which contributes to cellular decline. The activation of cyto-protective mechanisms, such as the cellular recycling process of autophagy and stress responses, contributes to improving cellular function and preventing aging-induced dysfunction. Discovering mechanisms that help maintain cellular integrity during aging, is therefore an important step towards developing new strategies for maintaining cellular homeostasis and organismal health, which could have great impact by increasing healthspan and eliminating age-related diseases.
“Cancer is one the main causes of death in the US. Our research is focused on understanding how we can harness the power of our immune system to attack and kill cancer cells and cure patients. I chose to join Sanford Burnham Prebys because of their collaborative research culture and state-of-the- art core facilities. I believe that teamwork is the foundation of scientific breakthroughs, and the Institute provides a perfect supportive and friendly environment to achieve this.”
Originally from the Netherlands, Kelly received her BS and MS from Utrecht University, and performed her PhD studies at the Netherlands Cancer Institute in Amsterdam working with Prof. Karin de Visser studying the role of the immune system in the metastatic spread of breast cancer. For her postdoctoral training, Kelly joined the lab of Prof. Max Krummel at the University of California San Francisco to study how tumor-associated myeloid cells affect anti-tumor T cell responses. Kelly is currently setting up her independent research team at Sanford Burnham Prebys.
Education
2016-2023: Postdoctoral Training, University of California San Francisco (Mentor: Prof. Max Krummel, focus on immune evasive cancer) 2017: PhD, Netherlands Cancer Institute Amsterdam/ Leiden University (Mentor: Prof. Karin de Visser, focus on role immune system in breast cancer metastasis, 2011-2016) 2011: MS, Biomedical Sciences, Utrecht University, Netherlands 2008: BS, Biology, Utrecht University, Netherlands
Honors and Recognition
2022: Selected Attendee for SITC Women in Cancer Immunotherapy Network (WIN) Leadership Institute 2022: Selected Attendee for Arthur and Sandra Irving Cancer Immunology Symposium 2022: Ray Owen Poster Award for Outstanding Poster Presentation at 60th Midwinter Conference for Immunologists at Asilomar (sponsored by AAI) 2020-2022: Parker Scholar Award awarded by the Parker Institute for Cancer Immunotherapy (PICI) 2018: Honorable Mention of poster presentation at UCSF/UCB/UCM Immunology Retreat 2018: Poster presentation ‘Excellence in Research Award’ awarded by the National Philanthropic Trust 2017-2019: NWO Rubicon postdoctoral fellowship awarded by the Netherlands Organization for Scientific Research (NWO) 2015: Best presentation award at the annual Tumor Cell Biology meeting of the Dutch Cancer Society (KWF) 2014: Travel scholarship awarded by the Dutch Foundation for Pharmacological Sciences (NSFW) 2010: Master scholarship awarded by the Dutch Cancer Society (KWF)
Memberships
2022-present: Society for Immunotherapy of Cancer (SITC) 2020-present: Parker Institute for Cancer Immunotherapy (PICI) 2012-present: American Association for Cancer Research (AACR)
Related Disease
Breast Cancer, Cancer Biology, Immune Disorders, Inflammation, Innate Immunity, Metastasis, Skin Cancer and Melanoma, Tumor Microenvironment, Tumorigenesis
Phenomena or Processes
Adaptive Immunity, Innate Immunity
Research Models
Mouse, Primary Cells
Techniques and Technologies
Cellular and Molecular Imaging
The Kersten lab studies the interactions between the immune system and cancer.
We are fascinated by the crosstalk between the immune system and cancer. Our goal is to understand how cancer cells hijack the normal physiology and function of immune cells to support tumor growth and evade destruction by the immune system.
Kelly Kersten’s Research Report
Cancer immunotherapy, harnessing a patient’s immune system to fight cancer, has revolutionized the way we treat cancer. However, a large proportion of patients do not respond to this type of therapy, and we do not fully understand why. In the Kersten lab, we study how different immune cells affect anti-tumor immunity with the ultimate goal to improve therapies to fight cancer. Research in our lab is focused on understanding how interactions between different immune cells in the tumor microenvironment, specifically macrophages and T cells, affect anti-tumor immunity and responsiveness to immunotherapy. Why do T cells become dysfunctional and exhausted? How do exhausted T cells modulate the composition of immune cells in tumors? And how do macrophages shut down anti-tumor T cells? Our research aims to define the molecular mechanisms that regulate these reciprocal signals to design novel anti-cancer therapies. How immune cells function is highly context-dependent. Upon infiltration in the tumor microenvironment, immune cells face extremely harsh conditions characterized by nutrient deprivation, hypoxia and metabolic challenges resulting in their failure to function properly. We study how different environmental factors impact immune cell phenotype and function, with the goal to optimize their cancer-killing properties.
Ten scientists at Sanford Burnham Prebys Medical Discovery Institute were awarded eight grants yesterday from Curebound, a San Diego-based philanthropic organization
Dr. Randal Kaufman previously served as professor of Biological Chemistry and Internal Medicine and Howard Hughes Medical Research Institute investigator at the University of Michigan Medical School. He received his PhD in pharmacology from Stanford University, where he studied gene amplification as a mechanism by which cells become resistant to anticancer agents. He was a Helen Hay Whitney fellow with Nobel Laureate Dr. Phillip Sharp at the Center for Cancer Research at the Massachusetts Institute of Technology (M.I.T.), where he developed gene transfer technologies based on gene amplification and expression in mammalian cells. He did his postdoctoral work at the Center for Cancer Research at M.I.T. In the 1980s, Dr. Kaufman’s experience with gene transfer and engineering led him to become a founding scientist at Genetics Institute Inc., where he engineered mammalian cells for high-level expression of therapeutic proteins, such as clotting factors that are now used to treat individuals with hemophilia. Dr. Kaufman joined Sanford Burnham Prebys in 2011.
Education
Postdoctoral, Center for Cancer Research, M.I.T. PhD, Stanford University BA, University of Colorado
Other Appointments
7/2011: Present Adjunct Professor, Department of Biological Chemistry, University of Michigan, Ann Arbor, MI
Honors and Recognition
2006: AAAS Fellow 2000: Distinguished Investigator Award-MI Hemophilia Society 1999: Investigator Recognition Award, International Society of Thrombosis and Haemostasis 1998: International Association Francaise Des Hemophiles Award 1993: Dr. Murray Thelin Award
Related Disease
Liver Diseases, Type 2 Diabetes
Phenomena or Processes
Protein Misfolding, The Unfolded Protein Response
The Kaufman lab is focused on understanding the fundamental mechanisms that regulate protein folding and the cellular responses to the accumulation of unfolded/misfolded proteins within the Endoplasmic Reticulum (ER). When proteins fail to fold correctly, they don’t work properly. More importantly, misfolded proteins accumulate with age and cause cellular toxicity, leading to almost every disease associated with aging. In many degenerative diseases, including neurological, metabolic, genetic, and inflammatory diseases, it’s thought that the accumulation of misfolded proteins leads to cellular dysfunction and death.
Dr. Kaufman’s research has focused for more than 30 years on mechanisms that regulate proper protein folding in the ER; this work contributed to the discovery of the UPR in the mid 1980s. The UPR pathways, mediated by PERK, IRE1, and ATF6, coordinate primarily an adaptive response. More recently, his research has focused on molecular mechanisms that establish the apoptotic program in response to protein misfolding in the ER, studies that have shed light on the mechanism by which cancer cells survive in a stressful environment.
Randal Kaufman’s Research Report
The major portion of our research is aimed at elucidating fundamental mechanisms that regulate protein folding and the cellular responses to the accumulation of unfolded protein within the (ER). Research into the fundamental processes that regulate protein synthesis and folding within the ER should have impact on the understanding of genetic diseases that result from protein folding defects.
Accumulation of unfolded/misfolded proteins within the ER induces an adaptive stress response known as the Unfolded Protein Response (UPR). The UPR signal is transduced from the ER lumen to cytoplasm and nucleus by three transmembrane proteins IRE1, ATF6, and PERK. UPR activation induces the expression of a family of basic leucine zipper-containing transcription factors that activate transcription of genes encoding functions to reduce the protein-folding load and increase the protein folding capacity of the ER. IRE1 is a serine/threonine protein kinase and endoribonuclease that signals transcriptional activation by initiating a novel splicing reaction on the mRNA encoding the transcription factor XBP1. UPR activation promotes trafficking of ATF6 from the ER to the Golgi where it is processed to yield a cytosolic fragment that is a potent transcriptional activator. In addition, the protein kinase PERK signals translational attenuation through phosphorylation of the alpha subunit of the eukaryotic translation initiation factor 2 (eIF2a) on serine residue 51. This phosphorylation attenuates translation of most cellular mRNAs but selectively induces translation of the transcription factor ATF4. We demonstrated that PERK/eIF2a signaling is essential for glucose-regulated insulin production by pancreatic beta cells, where defects in this pathway result in beta cell dysfunction and diabetes. The findings demonstrate an unprecedented link between glucose metabolism, mRNA translation, and protein folding and have implication in the treatment of diabetes. Future studies directed to elucidate the molecular logic for the UPR adaptive response will provide fundamental insight into numerous pathological conditions such as viral infection, cancer, inflammation, metabolic disease and atherosclerosis, and protein folding diseases such as Parkinson’s disease and Alzheimer’s disease.
 Oct 22, 2025
Oct 22, 2025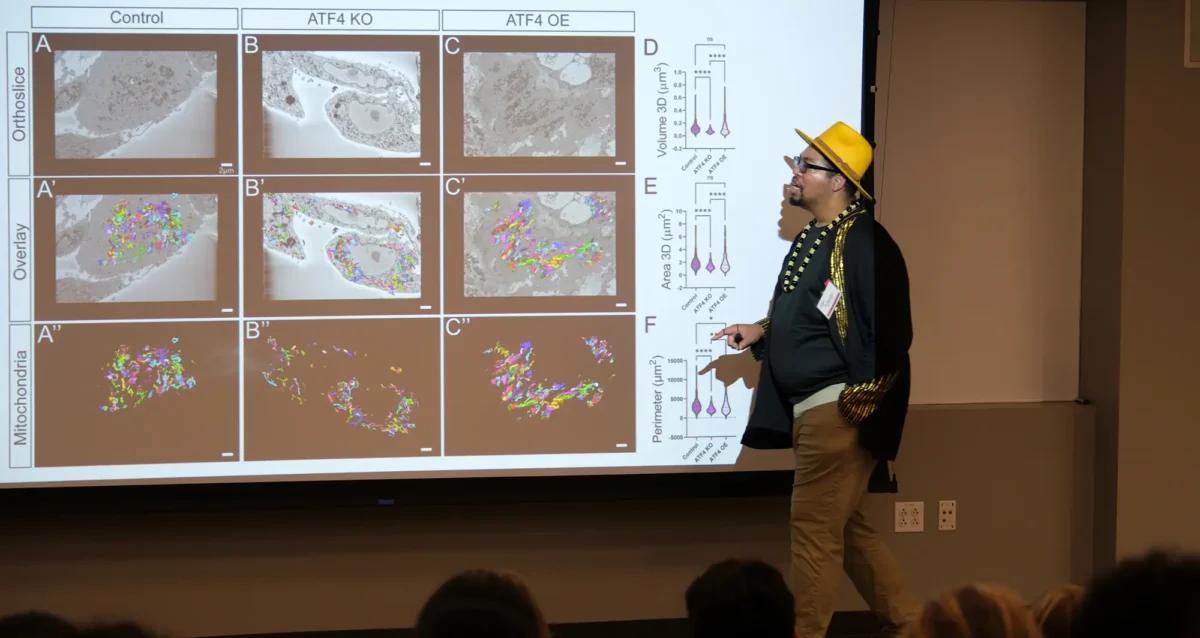 Sep 25, 2025
Sep 25, 2025 Aug 7, 2025
Aug 7, 2025 Jan 19, 2024
Jan 19, 2024 Nov 2, 2023
Nov 2, 2023 Aug 2, 2023
Aug 2, 2023