Dr. Brenner was named president and chief executive officer of Sanford Burnham Prebys in September 2022 after serving as vice chancellor for health sciences at UC San Diego and dean of its school of medicine for an unprecedented 15 years, during which he oversaw the launch and expansion of numerous multidisciplinary efforts, including the Institute for Engineering in Medicine, the Institute for Genomic Medicine, the Sanford Consortium for Regenerative Medicine, the UC San Diego Sanford Clinical Stem Cell Program, and the C3 Cancer Center Consortium (comprising UC San Diego, the Salk Institute for Biological Studies and Sanford Burnham Prebys).
Previously, he served as chair of the Department of Medicine and Physician-in-Chief of New York Presbyterian Hospital/Columbia University and, before that, as Chief of Gastroenterology and Hepatology at University of North Carolina at Chapel Hill.
As a distinguished physician-scientist, Brenner is a recognized leader in the field of gastroenterology research, with more than 200 peer-reviewed publications, two patents and ranking among Highly Cited Researchers by Web of Science and Clarivate Analytics.
He is an elected member of the National Academy of Medicine; past president of the Association of American Physicians and former editor of the journal Gastroenterology (2001 to 2006).
He is currently deputy editor of the journal PNAS Nexus.
Education and Training
1988: Fellowship, Gastroenterology, UC San Diego 1986: Fellowship, Genetics & Biochemistry, National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda, MD 1985: Intern & Residency, Internal Medicine, Yale University 1979: MD, Medicine, Yale University 1975: BS, Biology, Yale University
Awards, Honors and Recognition
2005: Fellow (FACP), American College of Physicians 1986: Bomedical Scholar, Pew Foundation
Other Appointments and Memberships
Alcoholic Beverage Medical Research Foundation American Society for Clinical Investigation Association of American Physicians American College of Physicians American Gastroenterological Association American Clinical and Climatological Association
Metabolic dysfunction–associated steatohepatitis, or MASH, is an inflammatory, liver-scarring disease that has reached epidemic proportions, with an estimated 1.5% to…
Dr. Zhang is professor and director of the Center for Neurologic Diseases at Sanford Burnham Prebys. Prior to that, he was professor and director of the Signature Program in Neuroscience & Behavioral Disorders at Duke-NUS Medical School, Singapore as well as professor of Neuroscience and Neurology, Steenbock Professor in Neural and Behavioral Sciences at the University of Wisconsin-Madison.
Dr. Zhang received his MD and MS in China and PhD in Canada. He is a pioneer in stem cells and regenerative medicine. He has developed technology to guide human stem cells to functionally specialized nerve cell types that are impaired in many neurological and psychiatric conditions with 25 awarded patents and several pending applications. He established the Stem Cell & Genome Editing Core at the UW-Madison and Duke-NUS, serving investigators on campus and beyond. He has also developed stem cell-based platforms for studying neural degeneration and testing drugs for neurological diseases. In parallel, he is developing cell therapy for neurological diseases like Parkinson’s disease, spinal cord injury and stroke. Dr. Zhang was a founding member of the WiCell Institute and co-founder of BrainXell, Inc and BrainXell Therapeutics, Inc.
Education
MD, Wenzhou Medical University, China MS, Shanghai Medical University, China PhD, University of Saskatchewan, Canada
Phenomena or Processes
Brain Aging, Neurodegeneration, Neuroregeneration
Anatomical Systems and Sites
Brain
Research Models
Human Pluripotent Stem Cells, Mouse, Rat
Techniques and Technologies
3D Bioprinting, Bioinformatics, Electrophysiology, Live Cell Imaging, Neural Circuit Tracing, Neural Transplantation, Rodent Behavioral Analysis, Stem Cell Differentiation and Engineering
The Zhang laboratory focuses on addressing how functionally diversified neuronal and glial subtypes are born in the building and rebuilding of our human brain. Over the past decades, they have developed models of neural differentiation from mouse, monkey, and human pluripotent stem cells, including embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs). Building upon the success in establishing transgenic and patient stem cells as well as directed neural differentiation to regionally and functionally specialized neuronal and glial subtypes, they are dissecting the cellular and molecular processes underlying brain aging and degeneration, focusing on motor neuron diseases (ALS, SMA), Alexander disease, Down syndrome, Parkinson’s disease and Alzheimer’s disease as well as some rare undiagnosed disorders. They are also transforming these cellular models to templates for drug discovery.
The Zhang laboratory has discovered that appropriately specified neurons project to correct brain regions and connect to the right targets in the adult rodent brain, suggesting a surprisingly regenerative capacity of human stem cell-produced neurons, very much like those born during embryonic development. They have also found that the transplanted human neurons receive appropriate inputs, a process largely dependent on the cell identity. They are currently evaluating the therapeutic potential of human stem cell-generated neural subtypes in animal (including non-human primate) models of Parkinson’s disease, stroke, and spinal cord injury. In particular, they have shown that cell therapy for Parkinson’s disease is safe and effective in a nonhuman primate model. They are now preparing for clinical trial of cell therapy for Parkinson’s disease.
Dr. Yip is professor and director of the Center for Data Science and Artificial Intelligence at Sanford Burnham Prebys Medical Discovery Institute. A leader in computational biology and bioinformatics at Chinese University of Hong Kong, he was recruited in 2022 to further elevate and accelerate Sanford Burnham Prebys’ growing capabilities and ambitions in next-generation biomedical research tools and approaches.
For almost 20 years, Dr. Yip’s research has focused on three primary interests: development of computational methods for analyzing data produced by emerging experimental technologies, such as single-cell and spatial transcriptomics; studying fundamental gene regulatory mechanisms using machine learning and data science methods; and identifying, annotating and interpreting genomic, transcriptomic and epigenomic changes in human diseases, such as cancers, diabetes, and neurodegenerative diseases.
Under his leadership, the mission of the Center for Data Science and Artificial Intelligence is to effectively tap the almost unlimited potential of rapidly evolving large-scale data sets and computational tools in biomedical research, with an emphasis on interdisciplinary collaborations that leverage the expertise of many disciplines to reveal new actionable knowledge.
Education and Training
2010: Postdoctoral associate, Molecular Biophysics and Biochemistry, Yale University 2009: PhD, Computer Science, Yale University 2003: M.Phil., Computer Science, The University of Hong Kong 1999: B.Eng., Computer Engineering, The University of Hong Kong
Related Disease
Biliary Atresia, Cancer, Diabetes – General, Hirschsprung Disease, Liver Cancer, Nasopharyngeal Carcinoma, Type 2 Diabetes
Phenomena or Processes
Cancer Epigenetics, Gene Regulation, Oncogenes, Posttranslational Modification, Transcriptional Regulation, Tumor Microenvironment
Anatomical Systems and Sites
Endocrine System, General Cell Biology, Immune System and Inflammation, Liver
Research Models
Computational Modeling
Techniques and Technologies
Bioinformatics, Comparative Genomics, Genomics, Machine Learning, Protein-Protein Interactions, Systems Biology
The Yip lab studies gene regulatory mechanisms by means of computational modeling. To facilitate their data-centric approach, they develop novel methods for analyzing large amounts of biological data, including those produced by cutting-edge high-throughput experiments. Their computational models provide a systematic way to investigate the functional effects of different types of perturbations to regulatory mechanisms, which creates testable hypotheses for studying human diseases and facilitates translational research.
Sanford Burnham Prebys scientists say that understanding the potential pitfalls of using artificial intelligence and computational biology techniques in biomedical…
Yu Yamaguchi earned his MD from Tohoku University in Japan in 1981, followed by a PhD in 1985, and training in obstetrics and gynecology at the same institute. Dr. Yamaguchi came to Sanford Burnham Prebys for his postdoctoral training. He was appointed to the staff in 1991.
Honors and Recognition
The Humanitarian Scientific Achievement Award, The MHE Research Foundation The Kushima Prize, The Alumni Association, Tohoku University School of Medicine
Related Disease
Alzheimer’s Disease, Arthritis, Autism Spectrum Disorders, Bone Mineralization Disorders, Epilepsy, Multiple Hereditary Exostoses
The goal of research in the Yamaguchi laboratory is to understand the role of proteoglycans and glycosaminoglycans in the context of development and human disorders. The general strategy is to define the role of proteoglycans and glycosaminoglycans by characterizing the phenotype of mutant mice lacking the synthesis of individual glycosaminoglycans. Specifically, mutant mice lacking the Ext1 and Has genes have been created to study heparan sulfate and hyaluronan, respectively. Recent progress in genetic studies in humans and mice has begun to reveal that deficiencies in glycosaminoglycans can be the causes and/or confounding factors of human childhood disorders. The Yamaguchi lab is now working to clarify the molecular mechanisms of two such disorders (multiple hereditary exostoses and autism) in order to develop new medical treatments.
For more information on the impact of Dr. Yamaguchi’s work, read letters from the patients with MHE.
Yu Yamaguchi’s Research Report
What Are Proteoglycans and Glycosaminoglycans?
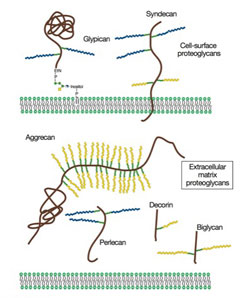
Proteoglycans are a family of glycoproteins consisting of a core protein and a various number of long sugar chains called glycosaminoglycans attached to the core protein (Fig. 1). There are four classes of glycosaminoglycans; heparan sulfate, chondroitin sulfate, keratan sulfate, and hyaluronan (hyaluronic acid). Heparin, the anticoagulant widely used in clinics, is a specialized form of heparan sulfate. Although there is ample circumstantial evidence that these glycosaminoglycans have important biological functions, a complete understanding of their function and their relevance to human diseases requires genetic animal models.
Figure 1. Proteoglycans consist of a protein core and one or more covalently attached glycosaminoglycan chains. From Esko, JD, Kimata, K., and Lindahl, U. Proteoglycans and Sulfated Glycosaminoglycans, In: Essentials of Glycobiology, CSH Press.
Developmental Roles Of Glycosaminoglycans
HEPARAN SULFATE – The Ext1 gene encodes an enzyme essential for the elongation of nascent heparan sulfate chains. As a result of genetic ablation of Ext1 using a ‘conditional knockout’ approach, heparan sulfate is eliminated from specific tissues and cell types. Our previous studies using brain-specific Ext1 conditional knockout have demonstrated critical roles of heparan sulfate in brain patterning, neurogenesis in the cerebral cortex, and pathfinding of various axon tracts (1)(2)(3)(4). More recently, conditional Ext1 knockout in developing limb bones revealed critical roles of heparan sulfate in the growth and patterning of bones and joint formation (5).
Moreover, our conditional Ext1 mutant mouse model has been distributed to more than 20 laboratories worldwide to characterize the role of heparan sulfate in various tissues and cell types, such as colon (6), kidney (7), lymphocytes (8), blood vessels (9), eyes (10), embryonic stem cells (11), and so on.
HYALURONAN – Three Has genes (Has1, Has2, Has3) encode the entire repertoire of hyaluronan synthases in mammalian cells. Genetic ablation of these genes, singly or in combination, results in a reduction or total elimination of hyaluronan, depending on the repertoire of Has expression in the given tissue. We created a conditional null allele of the Has2 gene, which is the predominant Has in many tissues. Our conditional Has2 knockout study targeted to the limb bud mesenchyme has revealed that hyaluronan plays a critical role in the proliferation and maturation of chondrocytes in the developing limb skeleton (12). Like Ext1 mutant mice, these Has2 conditional knockout mice are being used in more than a dozen laboratories worldwide for studies on the role of hyaluronan in various tissue and cell types.
Deficiencies in glycosaminoglycan synthesis can be the causes of childhood disorders
Recent progress in genetic studies in humans and mice has begun to reveal that deficiencies in glycosaminoglycans can be the causes and/or confounding factors of human childhood disorders. The research focus of our lab is to elucidate the molecular mechanisms of such disorders and to develop new medical treatments. We are currently studying two such disorders.
MULTIPLE HEREDITARY EXOSTOSES – One of the major diseases studied in our lab is Multiple Hereditary Exostoses (MHE; also known as Hereditary Multiple Exostoses [HME] or Multiple Osteochondroma [MO]). MHE is caused by a mutation in Ext1 (see above) or its related gene, Ext2. As mentioned above, these genes encode an enzyme necessary to produce heparan sulfate. MHE occurs in children of 0-12 years old. Although no comprehensive survey has been conducted, it is estimated that there are several thousand individuals affected by MHE in the US, which makes MHE one of the more prevalent among ’rare diseases’. Dr. Yamaguchi is a member of the scientific advisory board of the MHE Research Foundation and has been working to promote collaborations between basic scientists, academic physicians, and patient advocates.
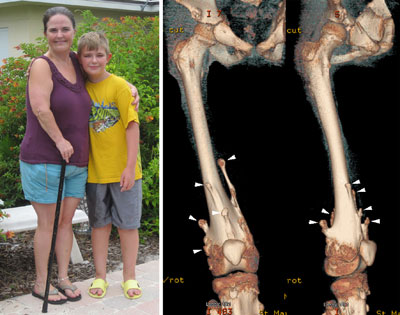
Figure 2. MHE patients, Carol and her 12-year-old son, Bruce. Shown on the right are three-dimensional CT images of Bruce’s right upper leg and knee joint area. Note that there are many bony protrusions (‘exostoses’), as indicated by white arrowheads. These tumors need to be surgically removed to prevent possible malignant transformation. Surgery is also needed to correct bone deformities and bone length inequalities. For example, Bruce and Carol have had 21 and 36 surgeries, respectively.
Children with MHE suffer from the formation of multiple –– sometimes as many as 100 –– bony tumors (osteochondromas) (Fig. 2). These bony tumors stunt their growth and can cause pain and disfigurement. Fortunately, the chance these tumors becomes cancerous is relatively low, partly because they are surgically removed as they develop. This means, however, children with MHE need to go through multiple surgeries over the course of their lives. There is currently no medical treatment for the disease.
Our lab is currently working to elucidate the molecular and cellular mechanisms of MHE. One of the major thrusts has been to create a mouse model that mimics the manifestations of human MHE. A long-term issue of MHE research has been the lack of mouse models that faithfully recapitulate the manifestation of human MHE; when Ext genes were inactivated in mice just as they are in human MHE patients, the mice failed to develop the symptoms of MHE.
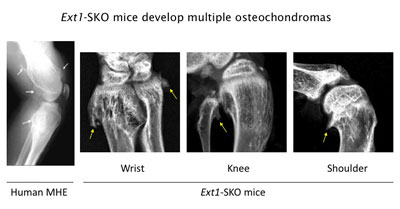
Figure 3. Ext1-SKO mice develop multiple osteochodromas in a pattern almost identical to human MHE. The X-ray images of the knee joint area of an MHE patient and the wrist, knee, and shoulder areas of Ext1-SKO mice. Osteochondromas are indicated by arrows. Ext1-SKO mice also mimic other skeletal deformities frequently seen human MHE, such as bowing of the forearm, the subluxation/dislocation of the radial head, and scoliosis (not shown).
Instead of knocking out the Ext1 gene in the whole mouse, we targeted the gene in only a small fraction of bone cells (Ext1-SKO mice). This minimalistic approach led to a mouse with all the physical manifestations of MHE, such as bony protrusions, short stature, and other skeletal deformities (Fig. 3)(13). The new mouse model answered some long-standing questions about MHE. Scientists had gone back and forth on whether osteochondromas observed in MHE are true tumors or just malformations of the bone. In this study, the tumors were made up of two cell types. A minority were mutant cells lacking Ext1, but, amazingly, most were normal bone cells. Hence, osteochondroma in MHE is not considered a true neoplasm in its strictest sense.
Our lab has been using this and additional mouse models to further dissect the pathogenic mechanism of MHE. Moreover, this mouse model provides new opportunities to test potential drugs to prevent osteochondroma formation and other clinical symptoms of MHE.
AUTISM – Children with MHE sometimes suffer from neurological and mental symptoms, which is not surprising because heparan sulfate is expressed and plays critical roles in the nervous system (1). The MHE community has long noticed the prevalence of autism-like behavioral traits in the patient population, and there are clinical reports describing the association of autism with MHE. Aided by the funds from the Sanford Health and the MHE Research Foundation, we have been studying the behavior of Ext1 mutant mice. Our preliminary data suggest that heparan sulfate has indeed a physiological function in the nervous system, and that its deficiency can cause behavioral deficits relevant to human autism. We are also analyzing DNA samples from individuals with autism for abnormalities in enzymes for heparan sulfate synthesis. Since heparan sulfate is a modulator of a number of neuronal molecules, we hope to identify functional networks of molecules underlying autism and other childhood mental disorders.
Kristiina Vuori earned her MD and PhD at University of Oulu, Finland. After completion of internship and residency, she received postdoctoral training at the Institute and was appointed to faculty in 1996. Dr. Vuori was selected as a PEW Scholar in the Biomedical Sciences in 1997. She has been co-Director of the Conrad Prebys Center for Chemical Genomics, housed at Sanford Burnham Prebys, since its inception in 2005. She was appointed Deputy Director of the Institute’s NCI-Designated Cancer Center in 2003, and Director of the Cancer Center in 2006. In 2008, she was appointed Executive Vice President for Scientific Affairs at Sanford Burnham Prebys. She was President of the Institute from 2010 to 2022.
Related Disease
Brain Cancer, Breast Cancer, Cancer, Leukemia/Lymphoma, Lung Cancer, Ovarian Cancer, Prostate Cancer
Dr. Vuori’s research is aimed at unraveling the cell mechanisms of the most life-threatening aspect of cancer, which is cancer metastasis. Metastasis is responsible for nearly all deaths in cancer patients, and understanding of the mechanisms that turn a cancer from a locally growing tumor into highly metastatic cancer cells will provide clues how to prevent this step in cancer progression. All cells in our body stick to one another and to the packaging material, or extracellular matrix, around them. This adhesion is essential for cell survival; if cells become detached from their microenvironment, they will die through a process known as apoptosis. This phenomenon, which is called adhesion dependency of survival, is one of the safeguards that maintain the integrity and normal function of tissues, and prevent cells from becoming cancerous. Normal cells cannot detach from their tissue and establish themselves somewhere else, because they will die on the way. Yet cancer cells somehow get around this requirement; they trespass aggressively into other tissues and metastasize to distant sites in the body without dying. Dr. Vuori’s work is aimed at identifying the molecular mechanisms that in normal cells makes them adhesion-dependent; false action of the very same mechanisms is likely to be the key step in allowing cancer cells to metastasize.
Evan Y. Snyder earned his MD and PhD (in neuroscience) from the University of Pennsylvania in 1980 as a member of NIH’s Medical Scientist Training Program (MSTP). He had also studied psychology and linguistics at the University of Oxford. After moving to Boston in 1980, he completed residencies in pediatrics and neurology as well as a clinical fellowship in Neonatal-Perinatal Medicine at Children’s Hospital-Boston, Harvard Medical School. He also served as Chief Resident in Medicine (1984-1985) and Chief Resident in Neurology (1987) at Children’s Hospital-Boston. In 1989, he became an attending physician in the Department of Pediatrics (Division of Newborn Medicine) and Department of Neurology at Children’s Hospital-Boston, Harvard Medical School. From 1985-1991, concurrent with his clinical activities, he conducted postdoctoral research as a fellow in the Department of Genetics, Harvard Medical School. In 1992, Dr. Snyder was appointed an instructor in neurology (neonatology) at Harvard Medical School and was promoted to assistant professor in 1996. He maintained lab spaces in both Children’s Hospital-Boston and at Harvard Institutes of Medicine/Beth-Israel Deaconess Medical Center. In 2003, Dr. Snyder was recruited to Sanford Burnham Prebys as Professor and Director of the Program in Stem Cell and Regenerative Biology. He then inaugurated the Stem Cell Research Center (serving as its founding director) and initiated the Southern California Stem Cell Consortium. Dr. Snyder is a Fellow of the American Academy of Pediatrics (FAAP). He also received training in Philosophy and Linguistics at Oxford University.
Related Disease
Alzheimer’s Disease, Amyotrophic Lateral Sclerosis (Lou Gehrig’s Disease), Arthritis, Brain Cancer, Brain Injury, Breast Cancer, Cancer, Childhood Diseases, Congenital Disorders of Glycosylation, HIV-Associated Dementia, Huntington’s Disease, Multiple Sclerosis, Muscular Dystrophy, Neurodegenerative and Neuromuscular Diseases, Neurological and Psychiatric Disorders, Parkinson’s Disease, Peripheral Vascular Disease, Skin Cancer and Melanoma, Spinal Cord Injury, Stroke, Traumatic Injury
We believe the study of stem cell biology will provide insights into many areas: developmental biology, homeostasis in the normal adult, and recovery from injury. Indeed, past and current research has already produced data in these areas that would have been difficult or impossible via any other vehicle. We have engaged in a multidisciplinary approach, simultaneously exploring the basic biology of stem cells, their role throughout the lifetime of an individual, as well as their therapeutic potential. Taken together, these bodies of knowledge will glean the greatest benefit for scientists and, most importantly, for patients. All of our research to date has been preformed in animal models with the ultimate goal of bringing them to clinical trials as soon as possible. Stem cells offer an intriguing mix of controversy, discovery, and hope. Politicians are charged with dealing with the controversial facets of stem cells, as we prefer to focus our energy on their potential for discovery and hope.
The Snyder Lab studies stem cell biology, with the goal of understanding normal development, tissue homeostasis, and recovery from injury and disease. A major focus is neural stem cells (NSCs), which can self-renew and differentiate into neurons, astrocytes, and oligodendrocytes. These properties make NSCs ideal for repair of damage due to injury or disease, but they also make them susceptible to transformation into malignant cancers.
Pier Lorenzo Puri earned his MD at the University of Rome “la Sapienza” in 1991. Dr. Puri completed his internship in Internal Medicine at the hospital “Policlinico Umberto I” (Rome) from 1992 to 1997, and defended an experimental thesis on the vascular effects of angiotensin II to graduate as Specialist in Internal medicine at the University of Rome “la Sapienza” in 1997. During this time he was frequently working at the Freien University of Berlin, as visiting scientist at the Deprtment of Biochemistry and Molecular Biology, to perform experiments of protein and DNA microinjection in cultured cells. Dr. Puri trained as a post-doctoral fellow at the University of California San Diego (UCSD), in the department of Cell Biology, under the supervision of Dr. Wang, from 1997 to 2001. He was appointed as Staff Scientist at the Salk Institute (La Jolla) in 2001, and became an Assistant Telethon Scientist at the Dulbecco Telethon Institute in Rome in 2002. He was upgraded to Associate Telethon Scientist at the Dulbecco Telethon Institute in Rome since 2007 and became Senior Telethon Scientist, Dulbecco Telethon Institute, in 2012, but declined this position. Dr. Puri joined Sanford Burnham Prebys as an Assistant Professor in 2004. He has been promoted to Associate Professor in 2010 and full Professor in 2015. From 2008 to 2016 Dr. Puri served as Adjunct Professor of Pediatrics at the University of California, San Diego. From 2008 to 2013 Dr Puri was an Associate Member of Sanford Children’s Health Research Center. Dr Puri has been Director of the laboratory of Epigenetics and Regeneration at Fondazione S. Lucia, Roma, Italy, but stepped down this position since 2019.
Education
University of California San Diego, Postdoctoral, Department of Biology University of Rome La Sapienza, PhD, Internal Medicine University of Rome La Sapienza, MD, Internal Medicine University of Rome La Sapienza, Undergraduate, Internal Medicine
Other Appointments
2020-2024: Member of the Science Advisory Board (SAB) European Commission-funded Consortium BIND (Brain Involvement In Dystrophinopathies) 2015-2019: Standing Member, NIH Study Section (SMEP) 2010-present: Member of Editorial Board of Skeletal Muscle
Phenomena or Processes
Adult/Multipotent Stem Cells, Aging, Cell Biology, Cell Cycle Progression, Cell Differentiation, Cell Signaling, Cellular Senescence, Development and Differentiation, Disease Therapies, DNA Damage Checkpoint Function, Epigenetics, Gene Regulation, Phosphorylation, Regenerative Biology, Signal Transduction, Transcriptional Regulation
Anatomical Systems and Sites
General Cell Biology, Musculoskeletal System
Research Models
Clinical and Transitional Research, Cultured Cell Lines, Human Adult/Somatic Stem Cells, Mouse Embryonic Stem Cells, Mouse Somatic Stem Cells, Primary Human Cells
Techniques and Technologies
Bioinformatics, Cellular and Molecular Imaging, Gene Expression, Genomics
Puri’s lab group investigates the molecular and epigenetic regulation of gene expression in skeletal muscle progenitors and other muscle-resident cell types (including fibro-adipogenic progenitors, cells from the inflammatory infiltrate, cellular components of neuro-muscular junctions) during physiological and pathological perturbations of skeletal muscle homeostasis.
We use molecular, biochemical and epigenetic tools to understand structural and functional principles of the 3D genome organization that regulates gene expression during muscle regeneration and diseases.
A topic of particular interest is the analysis of chromatin interactions that define the 3D genome organization and the identification of structural and functional interactions that regulate cell type-specific patterns of gene expression in response to cues released within the skeletal muscle regenerative environment in health and disease conditions, such as muscular dystrophies and other neuromuscular diseases.
The knowledge derived from our studies is instrumental to elucidate the pathogenesis of muscular disorders and discover pharmacological interventions that promote muscle regeneration to repair diseased muscles.
Current translational focus is devoted to:
the study of the therapeutic potential of HDAC inhibitors for treatment of Duchenne Muscular Dystrophy (DMD)
the identification of genome variants associated to DMD patient-specific patterns of expression of disease-modifier genes that can account for individual trends of disease progression beyond the common genetic deficiency of dystrophin
the effect of dystrophin deficiency and restoration by gene therapy on 3D genome and transcriptional output of DMD myofibers; the therapeutic potential of extracellular vesicles released by fibro-adipogenic progenitors of DMD skeletal muscles exposed to HDACi.
Puri Lab
Pier Lorenzo Puri’s Research Report
Puri’s lab group investigates the molecular and epigenetic regulation of gene expression in skeletal muscle progenitors and other muscle-resident cell types (including fibro-adipogenic progenitors, cells from the inflammatory infiltrate, cellular components of neuro-muscular junctions) during physiological and pathological perturbations of skeletal muscle homeostasis.
We use molecular, biochemical and epigenetic tools to understand structural and functional principles of the 3D genome organization that regulates gene expression during muscle regeneration and diseases
A topic of particular interest is the analysis of chromatin interactions that define the 3D genome organization and the identification of structural and functional interactions that regulate cell type-specific patterns of gene expression in response to cues released within the skeletal muscle regenerative environment in health and disease conditions, such as muscular dystrophies and other neuromuscular diseases.
The knowledge derived from our studies is instrumental to elucidate the pathogenesis of muscular disorders and discover pharmacological interventions that promote muscle regeneration to repair diseased muscles
Current translational focus is devoted to:
the study of the therapeutic potential of HDAC inhibitors for treatment of Duchenne Muscular Dystrophy (DMD)
the identification of genome variants associated to DMD patient-specific patterns of expression of disease-modifier genes that can account for individual trends of disease progression beyond the common genetic deficiency of dystrophin
the effect of dystrophin deficiency and restoration by gene therapy on 3D genome and transcriptional output of DMD myofibers; the therapeutic potential of extracellular vesicles released by fibro-adipogenic progenitors of DMD skeletal muscles exposed to HDACi.
1. Epigenetic regulation of skeletal myogenesis by histone acetyltransferases and deacetylases
Our earlier identification and characterization of acetyltransferases p300/CBP and PCAF, as transcriptional co-activators, and the histone deacetylases HDACs, as transcriptional co-repressors, of the myogenic determination factor MyoD1-3, respectively, inspired the experimental rationale toward exploiting pharmacological inhibition of HDAC to promote skeletal myogenesis.
2. HDAC inhibitors as pharmacological intervention in DMD and other muscular dystrophies
Puri lab discovered that dystrophin-activated nNOS signalling controls HDAC2 activity, thereby revealing a previously unrecognized link between constitutive activation of HDAC2 and alteration of the epigenetic landscape of dystrophin-deficient muscles6,7. This discovery established the rationale for using HDAC inhibitors to counter the progression of Duchenne muscular dystrophy (DMD), by correcting aberrant HDAC activity in dystrophin-deficient muscles8-11.
3. Control of chromatin structure in muscle cells by regeneration-induced signaling pathways
Upon the discovery and characterization of intracellular signaling pathways (i.e. p38, ERK and AKT cascades) that regulate muscle gene expression in myoblasts, in earlier studies during Puri’s postdoctoral training, Puri lab has revealed the mechanism by which muscle environmental cues are converted into epigenetic changes that regulate gene expression in healthy and diseased muscles, via extracellular signal-activated kinase targeting of chromatin-modifying enzymes. These studies provided the first evidence that regeneration activated p38 and AKT signaling cooperatively direct assembly and activation of histone acetyltransferases and chromatin remodeling SWI/SNF complex at myogenic loci in muscle progenitors12,13,15. Moreover, we discovered that regeneration-activated p38 targets Polycomb Repressory Complex (PCR2) at Pax7 locus to promote formation of repressive chromatin during satellite cells a ctivation14.
4. Epigenetic basis for activation of the myogenic program in ESCs and other pluripotent cell types
Puri lab studied the epigenetic determinants of human embryonic stem cells (hESCs) and induced pluripotent stem cells (hiPSCs) commitment to skeletal myogenesis, by investigating the hESC resistance to direct conversion into skeletal muscle upon ectopic expression of MyoD, which can otherwise reprogram somatic cells into the skeletal muscle lineage. These studies showed that hESC and hiPSC resistance to myogenic conversion is caused by the lack of expression of one structural component of the SWI/SNF chromatin remodelling complex – BAF60C – which is specifically induced in embryoid bodies13. Based on these studies, we have recently established a protocol of hESC-derived 3D contractile myospheres that offers the unprecedented opportunity to dissect and analyze the epigenetic dynamics that underlie the formation of skeletal muscles and to identify changes in the epigenome induced by contractile activity in healthy vs dystrophin-deficient myofibers16,20. We have also determined the identity of the general transcription factors implicated in the activation of skeletal myogenesis17, and we have discovered that replicative senescence is associated with acquisition of resistance to MYOD-mediated activation of muscle gene expression, caused by the constitutive activation of DNA damage repair (DDR) response that impairs cell cycle progression and MYOD activity18. Finally, our recent work has elucidated the mechanism by which MYOD regulates high-order chromatin interactions to define the tri-dimensional (3D) nuclear architecture for the activation of skeletal myogenesis during human somatic cell reprogramming into skeletal muscles19.
5. Identification, functional, phenotypic and molecular characterization of muscle-interstitial cells – (the fibroadipogenic progenitors – FAPs) in healthy and diseased muscles.
Our work has elucidated the molecular determinants of the interplay between adult muscle stem cells and cellular components of their functional niche (i.e. FAPs), by identifying regulatory networks implicated in compensatory or pathogenic regeneration, and suggesting “disease stage-specific” responses to pharmacological treatment of neuromuscular disorders, such as DMD. Indeed, we have shown that HDACi promote compensatory regeneration and prevent fibro-adipogenic degeneration in mdx mice at early stages of diseases, by targeting a population of muscle interstitial cells – FAPs8 – and have identified a HDAC-regulated network that controls expression of myomiRs and alternative incorporation of BAF60 variants into SWI/SNF complexes to direct the pro-myogenic or fibro-adipogenic FAP activity21. Furthermore, we have recently identified specific subpopulations of FAPs (subFAPs) in physiological conditions and disease22 and we have discovered that specific subFAPs expand and adopt pathogenic phenotypes upon muscle denervation23 or in muscles of patients affected by type 2 diabetes24.
Elena Pasquale earned her PhD in biology from the University of Parma, Italy. She did postdoctoral work at Cornell University, after which she was appointed Research Assistant Professor at University of Parma. Following a second postdoctoral training period at the University of California in San Diego, Dr. Pasquale was appointed Assistant Research Biologist at that institution. Dr. Pasquale was recruited to Sanford Burnham Prebys in 1990.
Related Disease
Cancer, Neurodegenerative and Neuromuscular Diseases, Skin Cancer and Melanoma
Cancer, Neurodegenerative and Neuromuscular Diseases
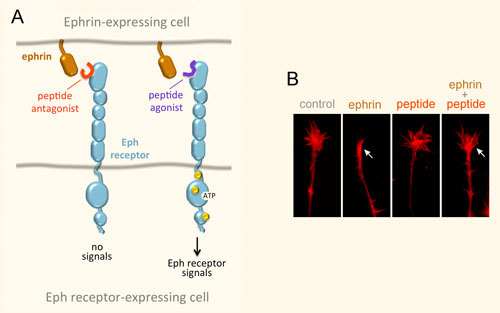
Receptor tyrosine kinases of the Eph family and their ligands, the ephrins, represent an important cell communication system that controls a vast array physiological and disease processes. For example, Eph receptors and ephrins take part in the formation of blood vessels, including the blood vessels that feed tumors, and regulate the malignant properties of cancer cells and their interplay with the tumor microenvironment. They also regulate the formation, plasticity and regeneration of neuronal circuits as well as neurodegenerative processes such as those occurring in amyotrophic lateral sclerosis (ALS) and Alzheimer’s disease. The signal transduction mechanisms of Eph receptors are intriguing, and complex, because these receptors engage in multiple modes of signaling. Binding to ephrin ligands on the surface of neighboring cells induces canonical signaling involving receptor clustering, autophosphorylation on tyrosine residues, and kinase activity-dependent downstream signaling. Binding to the Eph receptors can also cause the ephrins, which have a cytoplasmic domain or a GPI-anchor, to transmit signals. This leads to bidirectional signals emanating from Eph receptor-ephrin complexes positioned at sites of cell-cell contact. In addition, at least some Eph receptors can also signal through non-canonical mechanisms that are independent of ligand binding and kinase activity, for example through interplay with other receptor tyrosine kinase families and with serine/threonine kinases.
Our research investigates Eph receptor signaling activities in order to understand their role in normal physiology and in pathological conditions such as cancer and neurodegenerative disorders. This knowledge is useful for the development of disease treatments based on modulating Eph receptor/ephrin activities. Ongoing efforts in our laboratory also focus on the development of agents targeting Eph receptors for research and translational applications.
Elena Pasquale’s Research Report
We discovered several Eph receptors and ephrins, and research in our laboratory is dedicated to the characterization of Eph receptor signal transduction mechanisms and biological functions using biochemical, mass spectrometry, molecular biology and cell biology approaches in conjunction with animal models. We have identified tyrosine and serine/threonine phosphorylation sites of Eph receptors and ephrins using mass spectrometry and investigated the signaling role of these phosphorylation sites. For example, our past work showed that two conserved tyrosine phosphorylation sites in the juxtamembrane segment of the Eph receptors not only mediate association with binding partners but also regulate receptor kinase activity. We also found that the SRC and ABL non-receptor tyrosine kinases and the SHEP1 scaffolding protein are binding partners of the Eph receptors, and we identified signaling connections between Eph receptors and integrins. We also found that EphA4 is highly expressed in the adult brain, where it regulates synaptic connections. More recent work in our laboratory focuses on elucidating signaling pathways that mediate the activities of Eph receptors in cancer cells.
Tumor Suppression and Tumor Promotion by Eph Receptors
Many Eph receptors are highly expressed in tumors, but their role in cancer is incompletely understood and likely depends on the cellular context. Certain Eph receptors and ephrins promote tumor angiogenesis. We showed that the EphA2 receptor is upregulated in the tumor vasculature together with the ephrin-A1 ligand, which suggested a role in tumor angiogenesis that is now well established. We also found that the EphB4 receptor expressed on the surface of breast cancer cells can promote tumor xenograft growth by enhancing blood vessel formation through interactions with its preferred ligand, ephrin-B2, present in tumor endothelial cells. Additional intriguing roles for the Eph receptors in cancer progression have also emerged. We found that canonical signaling by the EphB4 receptor is low in breast cancer cells and that ephrin-induced stimulation of EphB4 kinase activity inhibits breast cancer cell malignancy in culture and tumor growth in vivo (Figure 1A) through inhibition of the CRK proto-oncogene. More recently, we elucidated an additional mechanism of tumor suppression mediated by canonical ephrin-induced EphA2 signaling (Figure 1A), which leads to inhibition of the AKT-mTORC1 oncogenic pathway through interplay of EphA2 with a phosphatase that dephosphorylates the AKT serine/threonine kinase.
Figure 1. Dual activities of Eph Receptors in Cancer Cells. (A) Eph receptor-ephrin binding at cell-cell contact sites results in the dimerization/clustering of Eph receptor-ephrin complexes, and initiation of canonical signals through the receptor cytoplasmic domain. Signals through the ephrins can also be generated. Tyrosine phosphorylation sites (yellow circles) promote Eph kinase activity and also provide binding sites for signaling proteins containing SH2 domains. Other effectors also mediate Eph signals, including PDZ domain-containing proteins. The Eph receptor domains are indicated on the left; LBD, ligand-binding domain. (B) Eph receptors can potentiate the oncogenic effects of other receptors. These activities are independent of ephrin binding and/or kinase activity and their mechanism is not well understood but in some cases depends on Eph receptor phosphorylation on serine/threonine residues (red circle).
There is also evidence that some Eph receptors can increase cancer cell malignancy through non-canonical ephrin-independent and/or kinase-independent signaling activities (Figure 1B), which is the subject of ongoing work. These tumor promoting activities include inducing invasiveness and metastasis, epithelial-to-mesenchymal transition, stem cell-like features and drug resistance.
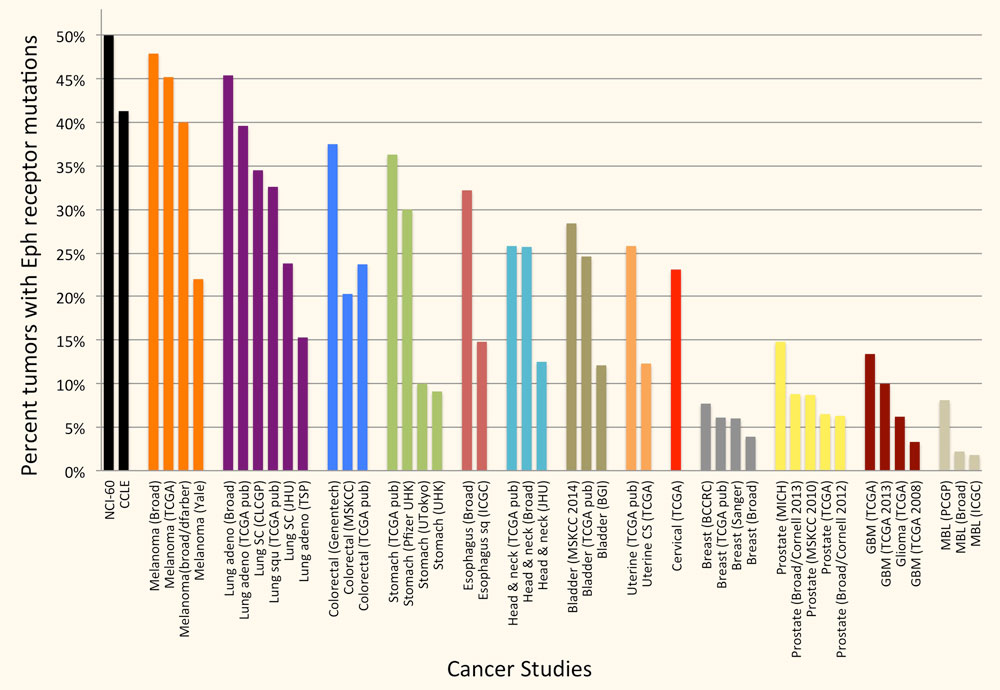
Eph Receptor Mutations in Cancer
The Eph receptors are frequently mutated in many types of cancer. In particular, genome-wide sequencing studies have detected somatic mutations in one or more Eph receptors in 25%-45% of melanomas, 15%-45% of lung cancers, 25-40% of colorectal cancers and 12%-25% of head and neck and uterine cancers (Figure 2), but very limited information is available on the effects of the mutations. Studies by ours and other groups have shown that a number of EphA2 and EphA3 mutations inactivate Eph receptor canonical signaling by disrupting ephrin binding or kinase activity, consistent with a role of canonical signaling in tumor suppression. Ongoing work in our laboratory focuses on characterizing the functional effects of Eph receptor mutations in cancers such as melanoma, and investigating whether the mutations shift the balance of the Eph receptor signaling activities from tumor suppression to tumor promotion. We are also interested in the interplay of Eph receptor mutations with mutations affecting well-established oncogenes and tumor suppressor genes. Understanding the effects of Eph receptor mutations in cancer cells will help shed light on the role of the Eph receptor/ephrin system in cancer cell transformation, malignant progression and drug resistance.
Figure 2. A large percentage of tumor specimens and cell lines harbor one or more Eph receptor mutations. Groups of bars of the same color represent studies of the same cancer type. The cancers with most Eph receptor mutations are shown; other tumor types have fewer or no Eph receptor mutations. The graph is based on data from cBioPortal for Cancer Genomics (www.cbioportal.org).
Peptides Targeting Eph Receptors
We have identified a number of peptides that bind to Eph receptors and inhibit ephrin binding by using phage display approaches. Collaborating groups have elucidated the structural features of several of these peptides in complex with the ligand-binding domain of Eph receptors, demonstrating that the peptides bind to the ephrin-binding pocket in the ligand-binding domain (Figure 3A). Most of the peptides are antagonists, but the peptides targeting EphA2 are agonists that activate receptor signaling and endocytosis similarly to the natural ephrin ligands. Interestingly, some of the identified peptides are highly specific and bind to only one Eph receptor family member. This is unlike the natural ephrin ligands, each of which promiscuously binds to multiple Eph receptors. Thus, Eph receptor-targeting peptides represent valuable pharmacological tools to study the functional importance of specific Eph receptors in tumors and the nervous system. Furthermore, they could be used as leads to develop therapies against cancer and neurological disorders, and to promote neural repair after nervous system injury (Figure 3B). Finally, our peptides have been used by other groups to deliver conjugated imaging agents, drugs and nanoparticles to Eph receptor-positive tumors. Current work focuses on identifying novel Eph receptor-targeting agents (such as peptides and small molecules) as well as improving the existing ones in collaboration with medicinal chemists and structural biologists, and evaluating them in cell culture and in vivo animal models.
Figure 3. Peptides can target the ephrin-binding pocket of Eph receptors with high affinity and specificity, affecting receptor function. (A) Peptides targeting the Eph receptors can function as antagonists that inhibit ephrin binding and receptor signaling, or in some cases as agonists that mimic the ephrins by activating Eph receptor signaling. Yellow circles indicate tyrosine phosphorylation sites in the activated Eph receptor. (B) An EphA4 peptide antagonist blocks ephrin-induced growth cone collapse in EphA4-expressing axons, suggesting its usefulness for promoting neural repair. The arrow in the second panel marks a growth cone collapsed due to ephrin treatment; the arrow in the fourth panel marks a growth cone that did not collapse following ephrin treatment in the presence of a peptide antagonist.
Andrei Osterman is a Professor in the Immunity and Pathogenesis Program Program at the Infectious and Inflammatory Disease Center of Sanford Burnham Prebys (since August 2003). He received his doctorate from Moscow State University in 1983, did postdoctoral work UT Southwestern Medical Center, and held the position of the Director and then Vice President of Research at Integrated Genomics in 1999-2003. Dr. Osterman is one of the founders of the Fellowship for Interpretation of Genomes (FIG), a nonprofit research organization that launched the Project to Annotate 1,000 Genomes in 2003. FIG provides the open-source integration of all publicly available genomes and tools for their comparative analysis, annotation, and metabolic reconstruction.
Related Disease
Breast Cancer, Cancer, Infectious Diseases, Radiation Damage, Skin Cancer and Melanoma
The main focus of Dr. Osterman’s research team is on fundamental and applied aspects of the key metabolic subsystems in a variety of species, from bacteria to human. This group uses a systems biology approach to reconstruct and explore metabolic and transcriptional regulatory networks. This approach combines comparative genomics and other bioinformatic techniques with biochemical and genetic experiments for pathway, gene and target discovery. Using this approach this group predicted and experimentally verified numerous enzyme families in the metabolism of cofactors, carbohydrates, and amino acids. Recent breakthroughs included prediction and characterization of novel transporters, transcriptional regulators and carbohydrate utilization pathways in a number of model bacterial systems. Applications in the field of infectious disease include identification of novel drug targets and structure-based development of novel anti-infective agents. New directions in cancer research are based on application of metabolic profiling technology for identification of novel diagnostic and therapeutic targets. Other directions of the on-going research include bioinformatics of regulatory proteolysis and applications of structural modeling for exploration of metabolic networks and gene discovery.
Sanford Burnham Prebys scientists say that understanding the potential pitfalls of using artificial intelligence and computational biology techniques in biomedical…
After receiving his early training in clinical chemistry/biochemistry at the University of Buenos Aires, Argentina, Dr. Millán first joined the La Jolla Cancer Research Foundation (LJCRF) in 1977, the predecessor of Sanford Burnham Prebys, as a trainee in clinical enzymology. He completed his PhD studies in Medical Biochemistry at the University of Umeå, Sweden and after post-doctoral stints in Copenhagen and LJCRF he was appointed to the faculty at SBP in 1986. He served as Professor of Medical Genetics in the Department of Medical Biosciences at his alma mater, Umeå University, Sweden, from 1995-2000. He was appointed Sanford Investigator at the Sanford Children’s Health Research Center at Sanford Burnham Prebys in 2008.
Honors and Recognition
2018: ASBMR Lawrence G. Raisz Award for Pre-clinical Research. 2001: Gold Medal of the Royal Academy of Medicine and Surgery, Murcia, Spain 1992: Honorary title of AcadémicoCorresponsal at the Royal Academy of Medicine and Surgery, Murcia, Spain.
Related Disease
Arthritis, Bone Mineralization Disorders, Cardiovascular Diseases, Colorectal Cancer, Crohn’s Disease (Colitis), Heart Disease, Inherited Disorders, Metabolic Syndrome, Peripheral Vascular Disease, Testicular Cancer
Phenomena or Processes
Cardiovascular Biology, Disease Therapies, Extracellular Matrix, Protein Structure-Function Relationships
Anatomical Systems and Sites
Cardiovascular System, Musculoskeletal System, Vasculature
Research Models
Mouse
The Millán laboratory works on understanding the mechanisms that control normal skeletal and dental mineralization and elucidating the pathophysiological abnormalities that lead to heritable soft bones conditions such as Hypophosphatasia (HPP) and to soft-tissue calcification, including vascular calcification, that is a hallmark in patients affected by a variety of rare genetic diseases as well as in chronic kidney disease. Dr. Millán’s research has already contributed to the implementation of a novel therapy for HPP, a genetic disease caused by deficiency in tissue-nonspecific alkaline phosphatase (TNAP) function, that leads to accumulation in the extracellular space of inorganic pyrophosphate (PPi), a potent inhibitor of mineralization. HPP is characterized by defective mineralization of bones (rickets or osteomalacia), and teeth that display a lack of acellular cementum, hypomineralized dentin and enamel, and periodontal defects. Dr. Millán’s team has demonstrated the effectiveness of enzyme replacement therapy using mineral-targeted recombinant TNAP (asfotase alfa) to prevent the skeletal and dental defects in the TNAP knockout mouse model of infantile HPP. This therapy was approved in 2015 for the treatment of patients with pediatric-onset HPP.
Current efforts, in collaboration with Professor Miyake’s group in Japan (https://www.nms-gt.org/en/members), focus on developing gene therapy as an alternative approach to treat HPP. Dr. Millán’s group has also identified key pathophysiological changes that lead to calcification of the arteries in animal models of generalized arterial calcification of infancy, pseudoxanthoma elasticum and related genetic diseases as well as in animal models of chronic kidney disease. His group, in collaboration with scientists at the Conrad Prebys Center for Chemical Genomics at Sanford Burnham Prebys, has developed proprietary compounds able to ameliorate the soft-tissue calcification in these conditions and clinical trials are now underway using these first-in-class small molecule inhibitors.