Evan Snyder, MD, PhD, professor in the Center for Neurologic Diseases, has been named a Fellow of the Child Neurology Society, honoring a distinguished career and significant contributions in the field of child neurology.
Snyder, who is also a member of the Cancer Genome and Epigenetics Program at Sanford Burnham Prebys, is a longtime leader in pediatric neuroscience and stem cell research. He joined Sanford Burnham Prebys in 2003, and was founding director of its original Stem Cell Research Center.
The Child Neurology Society is a professional organization whose mission is to promote the optimal care of children with neurological and developmental disorders by providing education, advocacy and support for clinicians and researchers in the field.
Snyder was cited for his clinical expertise, research contributions and ongoing commitment to the mission of the Society. He is featured in the second edition of Child Neurology: Its Origins, Founders, Growth and Evolution, a collection of biographies detailing the lives of innovators in child neurology throughout history.
Institute News
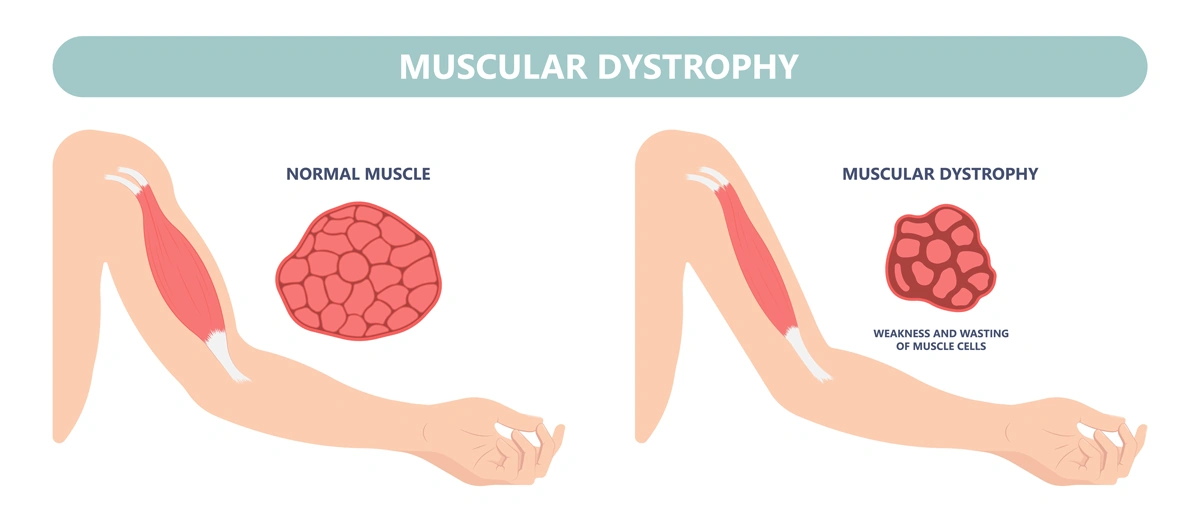
Decades of dedication led to FDA approval of a new treatment for Duchenne Muscular Dystrophy
Lorenzo Puri with his lab in Rome in 2004. From left: Lucia Latella, PhD; Silvia Fortuni, PhD; Christian Reale, PhD; Puri; Giulia Minetti, PhD; Cristiano Simone, PhD; and Carlo Serra, MD.
Nearly 30 years of discoveries by a Sanford Burnham Prebys scientist and collaborators lead to federal approval of the first non-steroidal drug to treat Duchenne muscular dystrophy.
For one San Diegan scientist at Sanford Burnham Prebys, the March 2024 federal approval of a new drug to treat Duchenne muscular dystrophy (DMD) marked a milestone in three decades of studying muscle regeneration and muscle-wasting diseases.
The compound, called Givinostat and marketed as DUVYZAT™, is a histone deacetylase inhibitor (HDACi)and was approved by the FDA for the treatment of boys with DMD.
“I have been working from the very beginning of my research career to translate early, basic discoveries into a treatment for DMD,” said Pier Lorenzo Puri, MD, director and professor in the Development, Aging and Regeneration Program at Sanford Burnham Prebys. “The lack of effective treatments for boys with DMD has left families and patients hopeless since the discovery of this disease. Witnessing the progression of such a disease without any option to counter its progression is cruel and I felt the urgency to help these people.”
DMD is the most frequent form of muscular dystrophy affecting approximately 1 in 3,500 male births. DMD is linked to the chromosome X, which harbors the gene coding the protein called dystrophin. As such, the disease develops only in males receiving from their mother the X chromosome carrying mutations in the dystrophin gene that impair production of dystrophin. Dystrophin is a protein that protects muscles from degeneration after they contract and relax. In its absence, the muscles of boys with DMD are prone to damage and undergo cycles of contraction and degeneration that eventually lead to muscle wasting and reduced function.
For decades, steroids have provided the standard-of-care for DMD, but steroids represent an empirical and palliative treatment based on their general anti-inflammatory properties, rather than a treatment that targets specific pathological events that contribute to DMD progression. Moreover, the chronic use of steroids is complicated by many side effects, including weight gain, weak bones, high blood pressure and behavior changes.
Puri decided to focus on the potential use of HDACi to treat DMD after he made seminal basic discoveries that revealed how muscle growth and regeneration are regulated by two enzymes with opposing activities: histone acetyltransferases and deacetylases.
“HDACi are not going to cure muscular dystrophy, but they do provide the first pharmacological treatment that can delay DMD progression, regardless of the type of mutation, and do so in a financially affordable way,” said Puri.
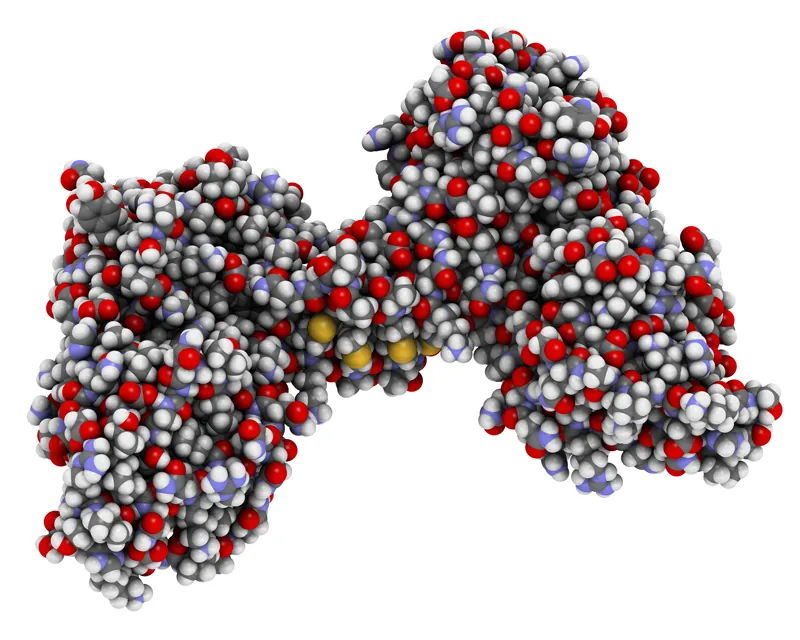
The molecular structure of dystrophin, a protein that protects muscles from degeneration after they contract and relax.
“It is important to note that FDA approval of Givinostat is not the end of the journey, but the beginning. The very good news here is that there is room to improve the efficacy of HDACi-based treatment for DMD by using already existing compounds or by developing novel molecules endowed with an improved therapeutic potential. This is because Givinostat has been used at sub-optimal concentrations due to potential adverse effects, and this might have limited its efficacy as a HDACi.
“People ask me whether Givinostat was approved because it is the most effective molecule among existing HDACi. The answer is that Givinostat has been the first, and so far the only, HDACi to be tested in clinical trials for boys with DMD. Givinostat might not entirely express the therapeutic potential of HDACi for DMD. A reasonable and exciting goal of future studies is to identify HDACi that surpass Givinostat in terms of therapeutic efficacy.”
Without dystrophin, the muscles of boys with Duchenne muscular dystrophy are prone to damage and undergo cycles of contraction and degeneration that eventually lead to muscle wasting and reduced function.
Puri noted that, “It is also important to carefully investigate functional interactions between HDACi and steroids, as the clinical trial with Givinostat has been performed while research participants were receiving steroids. However, because the activity of steroids is largely dependent on HDAC, it is very likely that these two treatments could collide, rather than synergize in producing beneficial effects on the muscles of DMD boys.”
He also emphasized that the impact of FDA approval of Givinostat extends way beyond the possibility to offer a treatment for DMD.
“It is the first evidence that it is possible to treat DMD by targeting pathogenic events induced as a consequence of dystrophin deficiency, as an effort parallel to gene and cell therapy, which will hopefully converge into future and more effective combined therapies.”
Building the basic science foundation
Puri’s contributions to the approval of the first non-steroidal drug to treat DMD span nearly 30 years of basic and preclinical research of diseases thought to be incurable — especially pediatric conditions he thought especially cruel.
The puzzle pieces began to take shape as Puri was studying the growth of muscle cells and skeletal muscle tissue, a biological process called skeletal myogenesis. He started working on this project in the 1990s in the laboratory of molecular biology directed by Massimo Levrero, MD, in Rome. Puri had earned his medical degree in 1991 before conducting an internship in Internal Medicine.
“I started there, in a lab inside a hospital in Rome,” said Puri. “I used to see patients until the early afternoon and then I was running to the lab to perform experiments. As a young clinician with a passion for basic research, I was always developing experiments with patients in mind.”
Puri decided to test his first hypothesis by using what was then an innovative technology called microinjection to insert DNA or antibodies inside cultured cells. He decided to spend several months at the Free University of Berlin in the laboratory of Adolf Graessmann, PhD, a pioneer of this technique. Puri later decided to go to the United States to work in the lab of Jean Y.J. Wang, PhD, at the University of California San Diego, to further develop his studies.
After entering the U.S., Puri has worked closely with his long-term collaborator and friend Vittorio Sartorelli, MD, currently the deputy scientific director of the National Institute of Arthritis and Musculoskeletal and Skin Diseases and chief of the institute’s Laboratory of Muscle Stem Cells & Gene Regulation. Together, they uncovered an important role for two groups of enzymes, histone acetyltransferases and deacetylases, that control access to DNA by altering the structure of chromatin.
Histone acetyltransferases control DNA accessibility by adding acetyl groups to histones, which loosens the wrapping of DNA around them, essentially ‘opening’ chromatin and promoting gene expression. Histone deacetylases reverse the process by removing acetyl groups, limiting the activity of genes and the production of key proteins.
“One of our seminal findings was the discovery of associations between myogenesis and an enzymatic activity that could be pharmacologically modulated,” explains Puri.
Puri with current members of hislab at Sanford Burnham Prebys.
Puri and Sartorelli started to explore the possibility that pharmacological modulators of this process by HDACi could affect the growth of muscle cell progenitors and their ability to form contractile muscle tissues.
After finding that HDACs limit muscle cell differentiation, the team’s next step was to find compounds (inhibitors) capable of blocking HDACs from removing acetyl molecules and reducing myogenesis. “We decided see if there is an HDAC inhibitor — an inhibitor of the inhibitor — because this could reduce muscle loss,” notes Puri. “There were a few compounds, and we found a strong effect every time we tested them.”
“I remember vividly the first time I saw the effect of HDACi on cultured muscle cells. I was a postdoc in Jean Wang’s lab at UC San Diego, and one morning I opened the incubator and found that muscle cells treated with HDACi had formed giant myotubes (the contractile muscle). Of course, we started to wonder whether such evidence could provide the rationale we were looking for. If so, this could pave the way toward discovering pharmacological interventions that may promote muscle regeneration.
Puri with Chiara Mozzetta, PhD, faculty at the Institute of Molecular Biology and Pathology at the National Research Council of Italy. Mozzetta previously conducted a postdoctoral fellowship in the Puri lab and made major contributions to the discovery of HDAC inhibitors as therapeutics for DMD.
“But, at that time, it sounded more like a dream. We did not even dare to imagine that the final outcome would have been the identification of a pharmacological treatment for DMD.”
Pursuing the preclinical potential
Puri and Sartorelli pursued their dream, driven by encouraging experimental evidence and discoveries. Still, it took years to provide the rationale for testing HDACi in DMD—years and a few fortunate coincidences. One was the identification of follistatin as a mediator of the action of HDACi on muscle cells. Follistatin is the endogenous inhibitor of myostatin, a potent inhibitor of muscle growth and size, ensuring that muscles do not grow too large.
It was during that time that other groups independently discovered that genetic mutations in the myostatin gene resulted in an abnormal increase in muscle mass in cattle, mice and humans. More importantly, it was published that genetic or pharmacological inactivation of myostatin could exert beneficial effects in a mouse model of DMD.
“That discovery suggested the rationale of testing whether HDACi could exert similar beneficial effect in the same mouse model of DMD,” said Puri. The team hypothesized that this could be achieved by using HDACi to block myostatin activity through the induction of follistatin.
Puri and Sartorelli decided to treat mdx mice — the mouse model of DMD — with a few HDACi. Puri performed these studies with a group of investigators that worked synergistically in his two labs (one in San Diego and the Dulbecco Telethon Institute laboratory in Rome) and in collaboration with Italian scientists Carlo Gaetano, PhD, and Claudia Colussi, PhD.
“The results of the experiment went beyond the most optimistic expectations,” said Puri. “The muscles were bigger. There was no scar tissue, no abnormal fatty deposits and less inflammation. The treated mice were running like normal mice.”
In follow-up studies, Puri and collaborators realized that the therapeutic properties of HDACi extended beyond the targeting of follistatin/myostatin interactions. The pace of discoveries increased along with the improved knowledge on DMD pathogenesis. Key and timely information was gained through identification of a population of muscle interstitial cells, called fibro-adipogenic progenitors (FAPs), from the laboratories of Fabio Rossi, MD, PhD, at the University of British Columbia in Canada and Dr. Kunihiro Tsuchida, MD, PhD, at Fujita Health University in Japan.
FAPs support muscle regeneration of normal muscles, but in dystrophin-deficient muscles these cells turn into the main effectors of fibrotic scars and fat infiltration — the most deleterious events in DMD pathogenesis. Studies from the Puri lab demonstrated that HDACi could restore the ability of FAPs from DMD muscles to promote regeneration, while blocking their pro-fibrotic and adipogenic activity.
Puri with collaborator Vittorio Sartorelli, MD, the deputy scientific director of the National Institute of Arthritis and Musculoskeletal and Skin Diseases.
“The identification of FAPs was a lucky coincidence, as it provided one of the main cell types targeted by HDACi and enabled the identification of the molecular mechanism of action that accounts for the therapeutic properties of HDACi,” said Puri.
Puri (at right) with collaborator Fabio Rossi, MD, PhD, professor of Medical Genetics at the University of British Columbia in Canada, at a muscle research meeting in Montrealin July 2024.
“It also helped to set in our preclinical studies the parameters that have been used in clinical trials. Indeed, I believe that one of the reasons for the success of this journey has been the development of a solid scientific rationale. I was definitely fortunate to have worked with a team of incredibly skilled young scientists that shared with me the wish to help DMD patients and the perseverance to take on a challenge for over 20 years. Overall, discovering the MOA of HDACs has been a fantastic journey.”
“Although the evidence that HDACi could be used to treat DMD was strong and the rationale was very solid, it was hard to convince big pharma to invest on this treatment. There were many excellent HDACi in the market that could have been used, but after knocking on many doors, no one was willing to partner on this task.
Puri felt compelled by these results to keep building support to convince potential industry partners to develop clinical trials. After talking with many businesses, he found Italfarmaco, an Italian pharmaceutical company with an HDAC inhibitor called Givinostat.
“In the end, I had to knock on the door of Italfarmaco, which owned Givinostat — an HDACi that I had never tested in my previous preclincial studies. This was another coincidence since Dr. Christian Steinkulher, a friend and colleague, alerted me about the potential availability of a pan-HDACi called Givinostat from Italfarmaco.
“When I approached Italfarmaco, I immediately realized that they were not very well prepared to take on this type of research. They were not familiar with DMD, and they didn’t have any previous experience on muscular diseases. They also had no background on the epigenetic effects of Givinostat.
“Until that time, they used Givinostat mostly for its anti-inflammatory properties, rather than the epigenetic regulation of gene expression. However, Italfarmaco recognized some potential in this operation and decided to give to me the opportunity to perform preclinical studies with Givinostat for DMD.”
Clinical trials and regulatory approval
After additional preclinical studies to better understand how Givinostat worked, Italfarmaco informed Puri that the company was ready to develop a clinical trial.
The results of the phase II clinical trial were published in Neuromuscular Disorders in 2016. During the trial, the scientists looked at whether the composition of the muscle improved. Muscle biopsies taken from 19 subjects who were treated for more than a year demonstrated that the drug had caused an increase in muscle fiber area and a decrease in fat deposition, scar tissue and other hallmarks of DMD.
“It was remarkable to see that the same positive effects observed in mdx mice treated with HDACi were also observed in DMD boys treated with Givinostat,” noted Puri. “The reproducibility of the outcomes in preclinical studies and clinical trials emphasizes the importance of performing accurate preclinical studies and identifying reliable outcome measures, as we did with HDACi for DMD.”
The trial also helped the team determine what dose of Givinostat was safe and still effective to use in the pivotal phase III trial that was reviewed by the FDA before the agency granted approval.
Italfarmaco established ITF Therapeutics in January 2024 as a new division that is now responsible for marketing DUVYZAT™ in the U.S. ITF Therapeutics announced that the drug was available in the U.S. on July 25. Italfarmaco’s Marketing Authorization Application (MAA) for Givinostat to the European Medicine Agency (EMA) was validated in fall 2023. This means that the drug is eligible to be reviewed by the EMA. If approved, Givinostat can be made available throughout the European Union (EU).
Puri surfs a wave in San Diego. Surfing is his passion outside of the laboratory.
Paving the way forward
Puri is not resting on his laurels and the approval of the first non-steroidal drug to treat DMD.
“Right now, we have steroids, HDAC inhibitors and gene therapy. We are working on the idea that gene therapy and HDAC inhibitors without steroids can perfectly synergize.”
Lorenzo and his dog Mojo.
The researchers are also investigating ways to enhance the effect of HDAC inhibitors through the use of extracellular vesicles (EV) released from FAPs following the exposure to HDACi. EVs are small biological bubbles that the body uses to carry compounds between cells. They are non-immunogenic and therefore suitable for transplantation into dystrophic muscles.
Puri is also investigating whether there are treatment conditions (including dietary supplements or other synergistic molecules) that can improve the therapeutic efficacy of HDACi. The researchers want to test if HDAC inhibitors can treat other forms of muscular dystrophy beyond DMD.
As much as Puri is focused on the future and continuing to find new and better approaches to treat muscular dystrophy, he also appreciates the importance of this vital moment and how the FDA’s decision positions the field for even more innovation.
“While muscular dystrophy was formally described by scientists 40 years ago, it has been a part of the human story since the beginning. People have been chasing something that could help, and for so long there was nothing to offer. Right now, we are paving the way for even better treatments that will be found.”
More information on the development of HDACi as a treatment for DMD is available in the following manuscripts:
Puri P.L., Avantaggiati M.L., Balsano C., San N., Graessmann A., Giordano A., and Levrero M. p300 is required for MyoD-dependant cell cycle arrest and muscle specific gene transcription. EMBO J. 16,369-383 (1997)
Puri P.L., Sartorelli V., Yang X.J., Hamamori Y., Ogrizko , Howard B., Kedes L, Wang J.Y.J., Graessmann A., Nakatani Y., Levrero M. Differential roles of p300 and PCAF acetyltransferases in muscle differentiation. Mol. Cell 1, 35-45 (1997)
Sartorelli V*., Puri P.L.* , Hamamori Y, Ogrizko V., Nakatani Y., Wang J.Y.J., Kedes L. Acetylation of MyoD directed by PCAF is necessary for the execution of the muscle program. Mol. Cell. 4, 725-734 (1999). *equal contribution
Puri P.L., Iezzi, S., Stiegler P., Chen T.T., Shiltz L., Muscat G., Giordano A, Wang J.Y.J. and Sartorelli V. Class I histone deacetylases sequentially interact with MyoD and pRb during skeletal myogenesis. Mol Cell. 8, 885-897 (2001)
Iezzi S., Cossu G., Nervi C. Sartorelli V., and Puri P.L. Stage-specific modulation of skeletal myogenesis by inhibitors of nuclear deacetylases Proc. Natl. Acad. Sci 99, 7757-7762 (2002)
Iezzi S., Di Padova M., Serra C., Caretti G., Simone C., Maklan E., Zhao P., Hoffman E., Puri P.L. and Sartorelli V. Deacetylase Inhibitors Increase Muscle cell Size by Promoting Myoblast Recruitment and Fusion Through Induction of Follistatin. Dev. Cell. 5:673-84. (2004).
Minetti G. C., Colussi c., Adami R., Serra C., Mozzetta C., Parente V., Illi B., Fortuni S., Straino S., Gallinari P., Steinkhuler C., Capogrossi M., Sartorelli V., Bottinelli R., Gaetano C., Puri P.L. Functional and morphological recovery of dystrophic muscles in mice treated with deacetylase inhibitors. Nature Medicine 12 (10): 1147-50 (2006)
Colussi C., Mozzetta C., Gurtner A. , Illi B., Straino S., Ragone G., Pescatori M., Zaccagnini G., Rosati G., Minetti G., Martelli F., Ricci E., Piaggio G., Gallinari P., Steinkulher C., Capogrossi M.C., Puri P.L*, Carlo Gaetano*. A Common Epigenetic Mechanism Underlies Nitric Oxide Donors and Histone Deacetylase Inhibitors Effect in Duchenne Muscular Dystrophy. Proc. Natl. Acad. Sci 105, 19183-7 (2008) *Corresponding authors. PMCID:PMC2614736.
Mozzetta C., Consalvi S., Saccone V., Tierney M., Diamantini A., Mitchel K.J., Marazzi G., Borsellino G., Battistini L., Sassoon D., Sacco A., Puri P.L. Fibroadipogenic progenitors mediate the ability of HDAC inhibitors to promote regeneration in dystrophic muscles of young, but not old mdx mice. EMBO Mol. Med. (2013), Apr;5(4):626-39 doi: 10.1002/emmm.201202096. [Epub ahead of print]. PMC Journal In Process.
Consalvi S, Mozzetta C, Bettica P, Germani M, Fiorentini F, Del Bene F, Rocchetti M, Leoni F, Mascagni P, Puri P.L., Saccone V. Preclinical studies in the mdx mouse model of Duchenne Muscular Dystrophy with the Histone Deacetylase inhibitor Givinostat. Mol Med. 2013 Mar 27. doi: 10.2119/molmed.2013.00011. [Epub ahead of print]. PMCID: PMC3667212.
Saccone V, Consalvi S, Giordani L, Mozzetta C, Barozzi I, Sandoná M, Ryan T, Rojas-Muñoz A, Madaro L, Fasanaro P, Borsellino G, De Bardi M, Frigè G, Termanini A, Sun X, Rossant J, Bruneau BG, Mercola M, Minucci S, Puri P.L. HDAC-regulated myomiRs control BAF60 variant exchange and direct the functional phenotype of fibro-adipogenic progenitors in dystrophic muscles. Genes & Development. 2014 Apr 15;28(8):841-57. doi: 10.1101/gad.234468.113. Epub 2014 Mar 28.
Bettica P, Petrini S, D’Oria V, D’Amico A, Catteruccia M, Pane M, Sivo S, Magri F, Brajkovic S, Messina S, Vita GL, Gatti B, Moggio M, Puri P.L., Rocchetti M, De Nicolao G, Vita G, Comi GP, Bertini E, Mercuri E. Histological effects of givinostat in boys with Duchenne muscular dystrophy.Neuromuscul Disord. 2016 Jul 11. pii: S0960-8966(16)30069-4. doi: 10.1016/j.nmd.2016.07.002. [Epub ahead of print]
Consalvi S, Tucciarone L, Macrì E, De Bardi M, Picozza M, Salvatori I, Renzini A, Valente S, Mai A, Moresi V, Puri P.L.. Determinants of epigenetic resistance to HDAC inhibitors in dystrophic fibro-adipogenic progenitors. EMBO Rep. 2022 Jun 7;23(6):e54721. doi: 10.15252/embr.202254721. Epub 2022 Apr 4. PMID: 35383427
Child malnutrition remains an alarming and appalling scourge.
In 2022, according to the World Health Organization, 148 million children in the world under 5 years were too short for their age (stunting) and another 45 million were too thin for their height (wasting) due to inadequate diet and nutrition.
Researchers around the world, including Andrei L. Osterman, PhD, professor in the Immunity and Pathogenesis Program at Sanford Burnham Prebys, have been investigating potential remedies, in particular some of the consequences of malnutrition, such as disturbed metabolism and immune/gut function.
In a new paper published October 2, 2024 in Science Translational Medicine, the multi-institutional team (including Osterman and colleagues at SBP) describe an interventional diet that essentially repairs the gut microbiome in children with moderate to severe acute malnutrition.
They conducted a three-month randomized controlled trial of a specialized food supplement in 12- to 18-month-old Bangladeshi children living in rural and urban slums with moderate acute malnutrition who had already been treated in hospital for severe acute malnutrition. The supplement, called microbiota-directed complementary food or MDCF-2, contains chickpea flour, peanut flour, soy flour, green banana, sugar, soybean oil and a vitamin-mineral premix, a formulation designed to promote the growth of therapeutic gut bacteria and improve the overall health and balance of the gut microbiome.
They found that MDCF-2 improved weight-for-age better than the traditional ready-to-use supplementary food (RUSF) used by relief agencies and others, which is composed of more traditional ingredients like rice, lentil, sugar, soybean oil and skimmed milk powder mixed with vitamins and minerals.
When excluding children unable to continue study participation due to severe flooding during the trial, the study authors also reported improvement of stunting at a faster rate. They tied these improvements in children’s health to Prevotella copri–associated metabolic changes.
P. copri (recently renamed as Segatella copri) is a bacterium found abundantly in the human gastrointestinal microbiome. Past studies have reported both positive and negative associations with health and disease. In the former, for example, healthy bacterial colonization of the gut has been positively correlated with conditions like inflammation, insulin resistance and diarrhea. It appears to be a major player in regulating dietary metabolism.
The bacterium is more common in non-Westernized populations consuming a diet rich in plants—the bacterium’s source of nutrients. In Western populations, it is associated with consumption of fruits and vegetables.
Genomic reconstruction of the metabolic potential of P. copri strains positively associated with infants’ health improvement confirmed their unique ability to utilize a large repertoire of plant-derived polysaccharides comprising MDCF-2 diet.
“This study can be viewed as a test of the generalizability of the efficacy and mechanism of action of MDCF-2 in acutely malnourished children,” said Osterman. “The main findings include the demonstration of significantly higher efficacy of MDCF-2 vs RUSF with respect to the improvement of (weight) growth.
“The success of the treatment was also manifest by beneficial changes in microbiome composition and by global changes of a range of serum protein biomarkers associated with healthy development.”
The findings, he said, also provide proof-of-concept that improving gut microbial health can be achieved using therapeutic nutrition and offers further guidance on how best to use microbiota-directed complementary foods.
Rapidly evolving computational tools may unlock vast archives of untapped clinical information—and help solve complex challenges confronting healthcare providers
The wealth of data stored in electronic medical records has long been considered a veritable treasure trove for scientists able to properly plumb its depths.
Emerging computational techniques and data management technologies are making this more possible, while also addressing complicated clinical research challenges, such as optimizing the design of clinical trials and quickly matching eligible patients most likely to benefit.
Scientists are also using new methods to find meaning in previously published studies and creating even larger, more accessible datasets.
“While we are deep in the hype cycle of artificial intelligence [AI] right now, the more important topic is data,” says Sanju Sinha, PhD, an assistant professor in the Cancer Molecular Therapeutics Program at Sanford Burnham Prebys. “Integrating data together in a clear, structured format and making it accessible to everyone is crucial to new discoveries in basic and clinical biomedical research.”
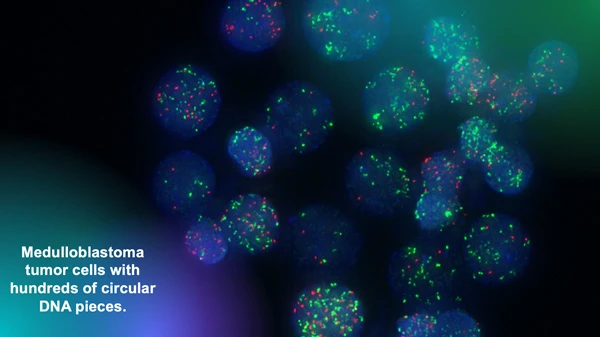
The Chavez lab uses fluorescent markers to observe circular extra-chromosomal DNA elements floating in cancer cells. Research has shown that these fragments of DNA are abundant in solid pediatric tumors and associated with poor clinical outcomes. Image courtesy of Lukas Chavez.
The Children’s Brain Tumor Network is another important repository for researchers studying pediatric brain cancer, such as Lukas Chavez, PhD, an assistant professor in the Cancer Genome and Epigenetics Program at Sanford Burnham Prebys.
“We have analyzed thousands of whole genome sequencing datasets that we were able to access in these invaluable collections and have identified all kinds of structural rearrangements and mutations,” says Chavez. “Our focus is on a very specific type of structural rearrangement called circular extra-chromosomal DNA elements.”
Circular extra-chromosomal DNA elements (ecDNA) are pieces of DNA that have broken off normal chromosomes and then been stitched together by DNA repair mechanisms. This phenomenon leads to circular DNA elements floating around in a cancer cell.
Sanju Sinha, PhD, is an assistant professor in the Cancer Molecular
Therapeutics Program at Sanford Burnham Prebys.
“We have shown that they are much more abundant in solid pediatric tumors than we previously thought,” adds Chavez. “And we have also shown that they are associated with very poor outcomes.”
To help translate this discovery for clinicians and their patients, Chavez is testing the use of deep learning AI algorithms to identify tumors with ecDNA by analyzing the biopsy slides that are routinely created by pathologists to diagnose brain cancer.
“We have already done the genomic analysis, and we are now turning our attention to the histopathological images to see how much of the genomic information can be predicted from these images,” says Chavez. “Our hope is that we can identify tumors that have ecDNA by evaluating the images without having to go through the genomic sequencing process.”
Currently, this approach serves only as a clinical biomarker of a challenging prognosis, but Chavez believes it can also be a diagnostic tool—and a game changer for patients.
“I’m optimistic that in the future we will have drugs that target these DNA circles and improve the therapeutic outcome of patients,” says Chavez.
“Once medicine catches up, we need to be able to find the patients and match them to the right medicine,” says Chavez. “We’re not there yet, but that’s the goal.”
Chavez is also advancing his work as scientific director of the Pediatric Neuro-Oncology Molecular Tumor Board at Rady Children’s Hospital in San Diego.
“Recently, it has been shown that new sequencing technologies coupled with machine learning tools make it possible to compress the time it takes to sequence and classify types of tumors from days or weeks to about 70 minutes,” says Chavez. “This is quick enough to take that technology into the operating room and use a surgical biopsy to classify a tumor.
“Then we could get feedback to the surgeon in real time so that more or less tissue can be removed depending on if it is a high- or low-grade tumor—and this could dramatically affect patient outcomes.
“When I talk to neurosurgeons, they are always in a pickle between trying to be aggressive to reduce recurrence risk or being conservative to preserve as much cognitive function and memory as possible for these patients.
“If the surgeon knows during surgery that it’s a tumor type that’s resistant to treatment versus one that responds very favorably to chemotherapy, radiation or other therapies, that will help in determining how to strike that surgical balance.”
Lukas Chavez, PhD, is an assistant professor in the Cancer Genome and Epigenetics Program at Sanford Burnham Prebys.
Artist’s rendering of X-shaped chromosomes floating in a cell alongside circular extra-chromosomal DNA elements.
Rady Children’s Hospital has also contributed to the future of genomic and computational medicine through BeginNGS, a pilot project to complement traditional newborn health screening with genomic sequencing that screens for approximately 400 genetic conditions.
“The idea is that if there is a newborn baby with a rare disease, their family often faces a very long odyssey before ever reaching a diagnosis,” says Chavez. “By sequencing newborns, this program has generated success stories, such as identifying genetic variants that have allowed the placement of a child on a specific diet to treat a metabolic disorder, and a child to receive a gene therapy to restore a functional immune system.”
Programming in a Petri Dish, an 8-part series
How artificial intelligence, machine learning and emerging computational technologies are changing biomedical research and the future of health care
Part 1 – Using machines to personalize patient care. Artificial intelligence and other computational techniques are aiding scientists and physicians in their quest to prescribe or create treatments for individuals rather than populations.
Part 2 – Objective omics. Although the hypothesis is a core concept in science, unbiased omics methods may reduce attachments to incorrect hypotheses that can reduce impartiality and slow progress.
Part 3 – Coding clinic. Rapidly evolving computational tools may unlock vast archives of untapped clinical information—and help solve complex challenges confronting health care providers.
Part 4 – Scripting their own futures. At Sanford Burnham Prebys Graduate School of Biomedical Sciences, students embrace computational methods to enhance their research careers.
Part 5 – Dodging AI and computational biology dangers. Sanford Burnham Prebys scientists say that understanding the potential pitfalls of using AI and other computational tools to guide biomedical research helps maximize benefits while minimizing concerns.
Part 7 – Simulating science or science fiction?By harnessing artificial intelligence and modern computing, scientists are simulating more complex biological, clinical and public health phenomena to accelerate discovery.
Part 8 – Acceleration by automation. Increases in the scale and pace of research and drug discovery are being made possible by robotic automation of time-consuming tasks that must be repeated with exhaustive exactness.
Institute News
At a symposium on rare diseases, smiles were in abundance
Our 2024 meeting this month was a cause for celebration. Partnering with the family support and information group, CDG CARE, the Sanford Children’s Health Research Center and sponsors invited scientists, families and physicians to share their stories – some technical, some heart-breaking, some updates of ongoing therapies and some describing new developments.
Five years ago, there were no therapies; now seven are moving into patients. Celebration indeed.
Our keynote speaker, Joni Rutter, PhD, director of the National Center for Advancing Translational Sciences, part of the National Institutes of Health, commented on our event: “Meetings that engage clinicians, scientists, advocates and families equally should be the standard. (Our) approach is a model of collaboration and impact.”
In this CDG CARE video of this month’s gathering, you can see the joy, optimism and hope that inspires and helps carry us all. Thanks to everyone for their support. It makes those smiles real.
Institute News
Sharing science and stories at Rare Disease Day Symposium
The Sanford Burnham Prebys Rare Disease Day Symposium brought patients, families, physicians, scientists, industry experts and advocates together with a focus on congenital disorders of glycosylation.
Sanford Burnham Prebys, in partnership with CDG CARE and the Sanford Children’s Health Research Center, hosted a Rare Disease Day Symposium in San Diego from March 1-3, 2024. The goal of the event was to share the latest scientific developments from researchers studying congenital disorders of glycosylation (CDG), and to foster new perspectives, ideas and collaborations to accelerate the creation and implementation of better therapies and treatment plans for those living with CDG.
CDG is an umbrella term for more than 190 disorders caused by mutations that impair glycosylation; the complex process by which cells build long sugar chains that attach to proteins called glycoproteins. CDG affects fewer than 2,000 children worldwide. When glycosylation is impaired, the sugar molecules on many of the body’s proteins are absent or incomplete, leading to serious, often fatal, malfunctions in various organ systems throughout the body.
Since 2010, Hudson Freeze, PhD, the William W. Ruch Distinguished Endowed Chair, professor and director of the Human Genetics Program, and director of the Sanford Children’s Health Research Center at Sanford Burnham Prebys, has organized an annual Rare Disease Symposium, where scientists, doctors and families gather from around the world to discuss the latest research and meet other families coping with rare diseases.
“At Sanford Burnham Prebys, we’re committed to rare disease research,” says David Brenner, MD, president, CEO and Donald Bren Chief Executive Chair at Sanford Burnham Prebys, during his welcoming remarks. “We believe we can make a unique contribution to society with this work, and in so doing make the world a better place.”
Brenner noted that Rare Research Day marked a time for academic medical centers across the US to celebrate the synergy between patients, families, physicians and scientists that is needed to advance research on all rare diseases, including CDG.
This was reflected throughout the symposium’s schedule and in the tenor of individual presentations. Patients and families were invited to give talks throughout the weekend to discuss the perspective of living with CDG or caring for a family member with CDG. The physicians and scientists who spoke consistently credited the patients and families for all they do to help raise funds and participate in research, including clinical trials that can add more appointments to already challenging calendars and routines.
The family reception on Friday, March 1, concluded the first day of the symposium with a more informal opportunity for patients, families, doctors and researchers to connect and socialize.
“This is the largest meeting we’ve ever had,” notes Freeze. “This gathering is an important part of nurturing the CDG research ecosystem by bringing experts together while also knitting us closer together with the people who really matter – the families.”
The symposium’s many sessions over three days included:
Friday, March 1
Scientific meeting
Introduction and welcome from Sanford Burnham Prebys president and CEO, David Brenner; Malin Burnham and Debra Turner, philanthropists and honorary trustees; and Congressional Representative Scott Peters from California’s 52nd Congressional District
Discussion of perspectives, challenges and triumphs led by parents, patients and advocates
Sessions on new therapies in development, the potential use of biotin as a treatment for many CDG patients, neurological disease, and gene therapy approaches, among others
Poster session
Family reception
Saturday, March 2
Scientific meeting
Keynote address on “Accelerating Treatment and Cures for Rare Diseases” from Joni Rutter, PhD, director of the National Center for Advancing Translational Sciences in the National Institutes of Health
Additional conversation about the experiences of parents and advocates
Sessions on clinical trial updates; especially strong were drug repurposing efforts leading to new and unexpected potential treatments
Doctor-is-in-session
Brought together medical researchers, clinicians, advocates, patients and their families for an afternoon of hands-on collaboration in small groups
Prior “Doctor-is-in-session” events have led to profound experiences and unlikely partnerships
Evening reception
Sunday, March 3
CDG CARE Scientific and Family Conference
CDG clinical care and management sessions included neurophysiology and epilepsy, growth charts and hormonal abnormalities, puberty and bone health
CDG research sessions included genetics 101, CDG updates, organoids as disease models and clinical trials as a partnership between physicians and patients, among others
CDG resource exchange sessions included educational planning and advocacy, speech and technology, therapy interventions, special needs planning and behavioral health and family planning
Institute News
Sanford Burnham Prebys research plays a key role in developing microbiome-directed complementary food to help save malnourished children
Among the consequences of childhood malnutrition is the underdevelopment of their gut microbiomes, critical to human health, from innate immunity to appetite and energy metabolism.
Although malnourished children gain some weight and grow better when fed a nutrient-rich diet, they do not catch up to their well-fed counterparts—and their gut microbiomes do not recover.
In a 2021 clinical trial, researchers at Washington University School of Medicine showed how a newly designed therapeutic food—a unique mix of peanuts, bananas and other foods dubbed microbiome-directed complementary food, or MDCF—more effectively nourished healthy gut microbial communities than standard dietary supplements.
Now, with bioinformatics support from Andrei L. Osterman, PhD, professor in the Immunity and Pathogenesis and Cancer Metabolism and Microenvironment programs at Sanford Burnham Prebys and his colleagues Dmitry Rodionov, PhD, and Alex Arzamasov, the multi-institutional scientific team has published new research that identifies and describes the bioactive elements of microbiome-directed food.
“These are naturally occurring carbohydrate structures that could, in theory, be recovered in large quantities from the by-product streams of food manufacturing and be used to produce prebiotics,” said senior study author Jeffrey I. Gordon, MD, the Dr. Robert J. Glaser Distinguished University Professor at Washington University.
“We also have identified the microbes that process these food components, and in theory, there is potential for the organisms themselves to be part of a therapeutic intervention in children completely lacking these beneficial gut microbes.”
Osterman’s lab contributed bioinformatics analyses of 1,000 new metagenomically assembled genomes, or MAGs, representing the gut microbiomes of healthy Bangladeshi infants. The analyses included genome-based inference of the presence or absence in these MAGs of functional metabolic pathways for 106 major nutrients and intermediary metabolites.
“These predictions enabled the assessment of the microbiome-wide representation or enrichment of dietary carbohydrate utilization capabilities across numerous biospecimens from a randomized, controlled trial of MDCF in Bangladeshi children with moderate acute malnutrition,” said Osterman.
“The analyses helped elucidate glycan components of MDCF metabolized by bacterial taxa that are positively associated with healthy weight growth. The knowledge will help guide the therapeutic use of current MDCF and enable development of new formulations.”
Childhood undernutrition is a global scourge. In 2020, an estimated 149 million children under the age of 5 had stunted growth (low height for age), and 45 million exhibited stunting (low weight for height). More than 30 million children worldwide have moderate, acute malnutrition.
Undernutrition and its consequences are the leading causes of disease and death for children in this age range. An estimated 3 million children die each year due to poor nutrition and hunger.
Institute News
Hudson Freeze joins experts to discuss testing to help CAD-affected children
Hudson Freeze joined an international panel of genetics experts on CAD deficiency: Beyond the genetics—a podcast offered by the Journal of Inherited Metabolic Disease.
The researchers shared how clinical and functional genomics tests can accelerate the diagnosis of CAD-deficient patients and enable their timely treatment with uridine, a nutritional supplement that has dramatically improved the lives of other children with the condition.
“The effect of uridine for some children with CAD deficiency is nothing short of amazing. These kids go from bedridden to interacting with people and moving around,” says Freeze, PhD, director of the Human Genetics Program at Sanford Burnham Prebys.
CAD deficiency is a congenital disorder of glycosylation (CDG), an umbrella term for more than 170 disorders caused by mutations that impair glycosylation; the complex process by which cells build long sugar chains that attach to proteins called glycoproteins.
These tests allow us to identify CAD genetic variants, and to help affected children get the best treatment possible,” adds Freeze.
Institute News
Where science meets patients: Sanford Children’s Research Center hosts inaugural symposium
The event celebrated 16 years of progress at the Center and connected scientists with the people most impacted by their work.
The Sanford Children’s Health Research Center at Sanford Burnham Prebys recently hosted its first-ever Children’s Health Research Symposium, which brought scientists and families together to learn about the latest research tackling childhood diseases.
“We’re all here because we want to improve the health of children,” said President and CEO David A. Brenner, MD, during his opening comments. “But this event also shows the amazing amount of collaboration and collegiality across San Diego, because we have all types of people together from different backgrounds who want to develop therapies and cures for children affected by disease.”
The Sanford Children’s Health Research Center was established in 2008 with the help of a generous gift from Institute namesake T. Denny Sanford. Since then, the Center has been a world leader in children’s health research.
“T. Denny Sanford made an investment in children’s health 15 years ago, and I think that investment has paid off pretty well so far,” said Center director Hudson Freeze, PhD, in his introduction to the first scientific session. Freeze is among the world’s leading experts on congenital disorders of glycosylation (CDG), a rare group of genetic disorders that can cause serious, sometimes fatal, malfunctions of different organs and systems in the body.
“We’ve published over 600 scientific papers, and about half of those are translational studies, which means they’re helping turn scientific discoveries into real treatments for patients,” adds Freeze.
Professor Hudson Freeze with the Omler family
The day included presentations from researchers at Sanford Burnham Prebys, as well as from other research organizations studying childhood diseases. However, the highlight of the event was the afternoon reception, in which scientists had the opportunity to mingle and share a meal with families affected by rare childhood diseases.
Professor José Luis Millán (center) with the Fischer family (left) and the Britt family (right)
Each researcher briefly introduced the family affected by the illness the scientist studies. This list included many longtime friends of the Institute, such as Damian Omler, who lives with a rare form of CDG; and Morgan Fischer, who was born with soft bone disease. Today, thanks to the help of a drug developed based on the research of Institute professor José Luis Millán, PhD, Morgan is a thriving teenager. This drug is also helping other children living with soft bone disease, including 10-year-old Aubrey Britt, who was in attendance with her family as well.
“Something so important that we keep as a tradition for scientific events at our Institute is to involve families that have been touched by the work of our faculty,” said Freeze. “They’re why we’re all here.”
The full list of talks included:
Sanford Children’s Health Research Center
José Luis Millán, PhD “Developing therapeutics for soft bones and ectopic calcification disorders”
Duc Dong, PhD “From hope for few to drug for many—why rare is precious”
Evan Snyder, MD PhD “A clinical trial using human neural stem cells for neuroprotection in perinatal asphyxia, a major cause of cerebral palsy in kids”
Anne Bang, PhD “Drug screens of human-induced pluripotent stem cell (hiPSC) derived neuronal networks on multi-electrode arrays”
Pamela Itkin-Ansari, PhD “Proinsulin misfolding in diabetes”
Yu Yamaguchi, MD PhD “Multiple hereditary exostoses—from genetics to potential drug targets”
As an assistant professor at Sanford Burnham Prebys and director of the Neuro-Oncology Molecular Tumor Board at Rady Children’s Hospital, Lukas Chavez, PhD, leverages modern technology for precision diagnostics and for uncovering new treatment options for the most aggressive childhood brain cancers.
We spoke to Chavez about his work and asked him how modern technology—particularly cloud computing—is shifting the approach to cancer research.
How are you using new technologies to advance your research?
New technologies are helping us generate a huge amount of data as well as many new types of data. All this new information at our disposal has created a pressing need for tools to make sense of it and maximize their benefits. That’s where computational biology and bioinformatics come into play. The childhood brain cancers I work on are very rare, which has historically made it difficult to study large numbers of cases and identify patterns.
Now, data for thousands of cases can be stored in the cloud. By creating data analysis tools, we can reveal insights that we would never have seen otherwise. For example, we’ve developed tools that can use patient data in the cloud to categorize brain cancers into subtypes we’ve never identified before, and we’re learning that there are many more types of brain tumors than we’ve previously understood. We’re basically transforming the classic histo-pathological approach that people have studied for decades by looking at tumor tissues under the microscope and turning that into data science.
How is cloud computing improving cancer research in general?
Assembling big datasets delays everything, so I believe the main idea of cloud computing is really to store data in the cloud, then bring the computational tools to the data, not the other way around.
My team did one study where we assembled publicly available data, and basically downloaded everything locally. The data assembly process alone took at least two to three years because of all the data access agreements and legal offices that were involved.
And that is the burden that cloud computing infrastructures remove. All of this personalized cancer data can be centrally stored in the cloud, which makes it available to more researchers while keeping it secure to protect patient privacy. Researchers can get access without downloading the data, so they are not responsible for data protection anymore. It’s both faster and more secure to just bring your tools to the data.
Are there any risks we need to be aware of?
Like any new technology, we need to be clear about how we use it. The technology is another tool in the toolbox of patient care. It will never entirely replace physicians and researchers, but it can complement and assist them.
Also, because we use costly and sophisticated tools that are being built and trained on very specific patient groups, we need to be careful that these tools are not only helping wealthier segments of society. Ideally, these tools will be expanded worldwide to help everybody affected by cancer.